Scanpy单细胞处理流程之二:数据预处理
待补充: 1:详细的解释 2:图片美化的技巧
#安装导入需要的模块
import numpy as np
import pandas as pd
import scanpy as sc
import os
import torch
import omicverse
import matplotlib.pyplot as plt
/home/zhaozm/anaconda3/envs/sccell/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_csv from `anndata` is deprecated. Import anndata.io.read_csv instead.
warnings.warn(msg, FutureWarning)
/home/zhaozm/anaconda3/envs/sccell/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_excel from `anndata` is deprecated. Import anndata.io.read_excel instead.
warnings.warn(msg, FutureWarning)
/home/zhaozm/anaconda3/envs/sccell/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_hdf from `anndata` is deprecated. Import anndata.io.read_hdf instead.
warnings.warn(msg, FutureWarning)
/home/zhaozm/anaconda3/envs/sccell/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_loom from `anndata` is deprecated. Import anndata.io.read_loom instead.
warnings.warn(msg, FutureWarning)
/home/zhaozm/anaconda3/envs/sccell/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_mtx from `anndata` is deprecated. Import anndata.io.read_mtx instead.
warnings.warn(msg, FutureWarning)
/home/zhaozm/anaconda3/envs/sccell/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_text from `anndata` is deprecated. Import anndata.io.read_text instead.
warnings.warn(msg, FutureWarning)
/home/zhaozm/anaconda3/envs/sccell/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_umi_tools from `anndata` is deprecated. Import anndata.io.read_umi_tools instead.
warnings.warn(msg, FutureWarning)
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
Version: 1.6.2, Tutorials: https://omicverse.readthedocs.io/
# 检查当前的工作路径
current_working_directory = os.getcwd()
print(f"当前的工作路径: {current_working_directory}")
# 检查 torch 的 GPU 版本
if torch.cuda.is_available():
print(f"torch GPU 版本: {torch.version.cuda}")
else:
print("未检测到 GPU")
sc.settings.verbosity = 3# 日志详细程度
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80,facecolor='white')
当前的工作路径: /mnt/DEV_8T/zhaozm/scanpy_单细胞流程/pbmc_test
torch GPU 版本: 12.1
scanpy==1.10.1 anndata==0.11.1 umap==0.5.6 numpy==1.26.4 scipy==1.11.4 pandas==2.2.3 scikit-learn==1.5.0 statsmodels==0.14.2 igraph==0.11.5 pynndescent==0.5.13
## 读取文件
adata = sc.read_h5ad('pbmc3k.h5ad')
adata
AnnData object with n_obs × n_vars = 2700 × 32738
var: 'gene_ids'
# 全局设置背景
plt.rcParams["figure.facecolor"] = "white"
plt.rcParams["savefig.facecolor"] = "white"
# 设置全局图例文字颜色
plt.rcParams["legend.labelcolor"] = "black"
plt.rcParams["text.color"] = "black"
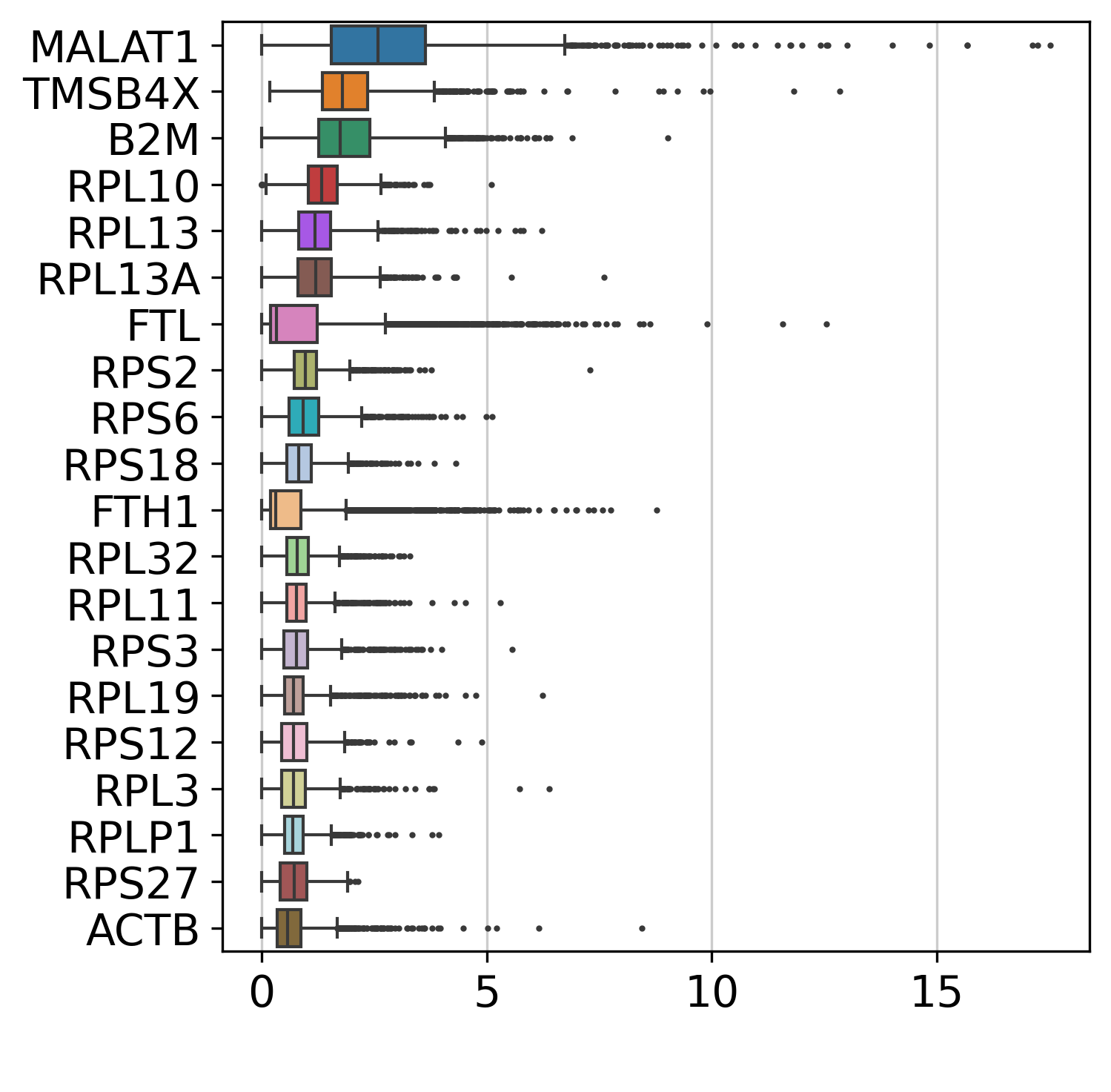
## 查看高变基因
sc.pl.highest_expr_genes(adata, n_top=20,show=False)
plt.savefig("./fig2/hvgmarker_plot.png", bbox_inches="tight", dpi=300)
normalizing counts per cell
finished (0:00:00)
## 筛选最少表达的基因和细胞
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
filtered out 19024 genes that are detected in less than 3 cells
## 计算线粒体基因
adata.var["mt"] = adata.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True
)
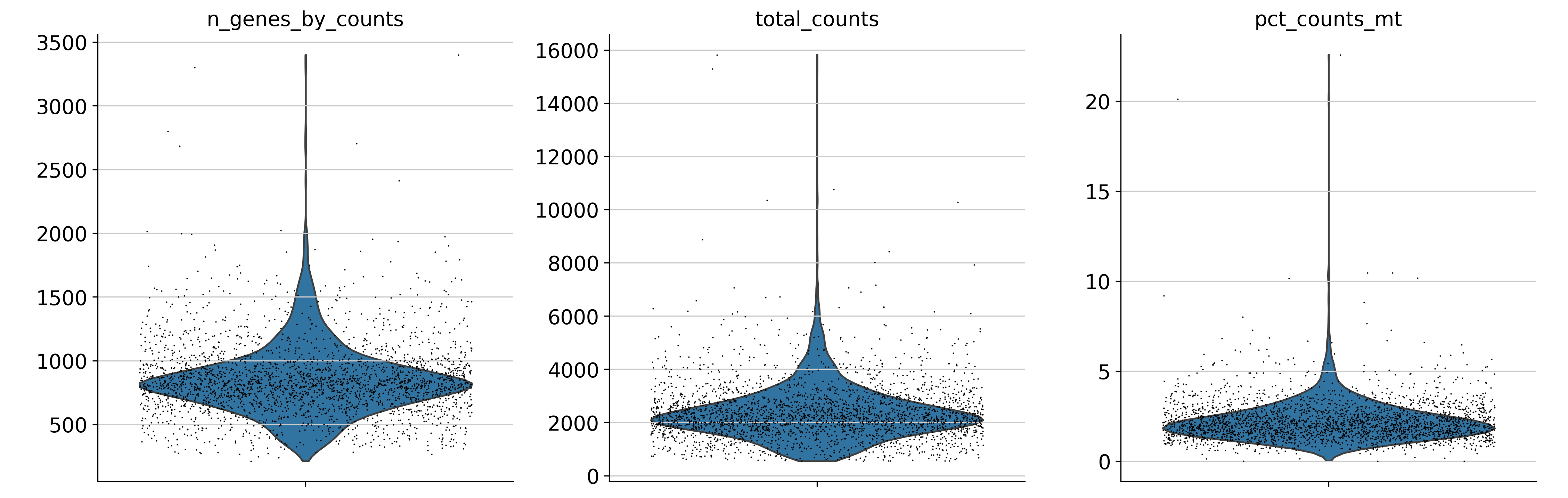
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
show=False)
plt.savefig("./fig2/vio_before_plot.png", bbox_inches="tight", dpi=300)
# 核糖体基因
adata.var["ribo"] = adata.var_names.str.startswith(("RPS", "RPL"))
# 血红蛋白基因
adata.var["hb"] = adata.var_names.str.contains(("^HB[^(P)]"))
## 通过切片的方式去除相应的细胞
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :].copy()
adata
AnnData object with n_obs × n_vars = 2638 × 13714
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'ribo', 'hb'
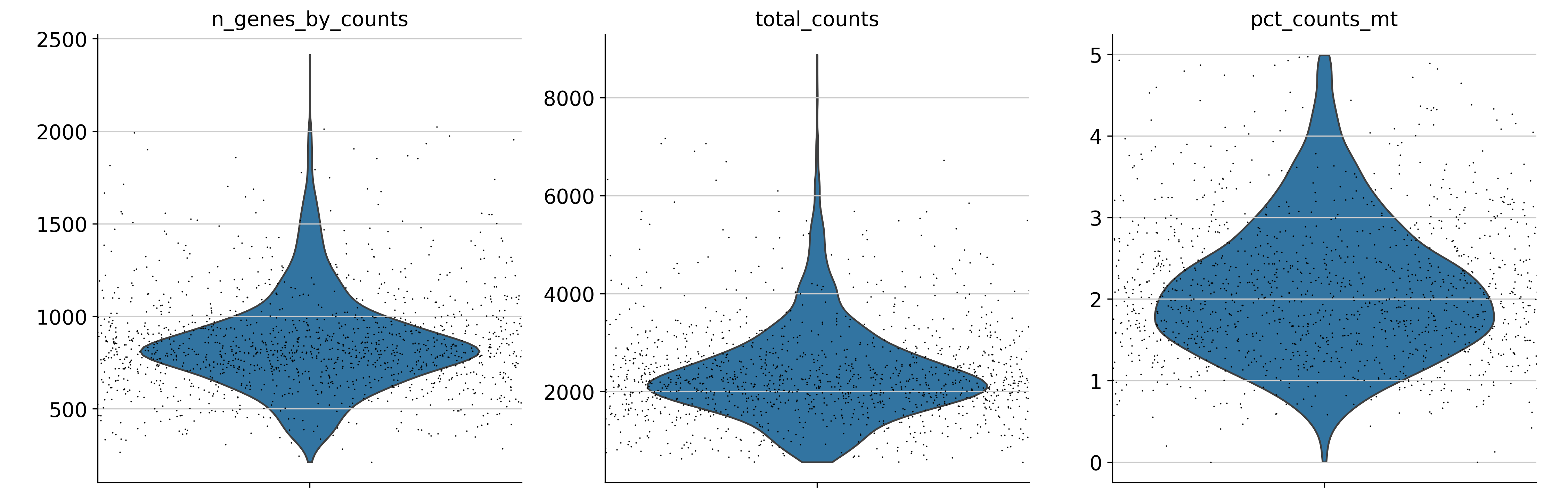
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=1,
multi_panel=True,
show=False)
plt.savefig("./fig2/vio_after_plot.png", bbox_inches="tight", dpi=300)
## 标准化
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
normalizing counts per cell
finished (0:00:00)
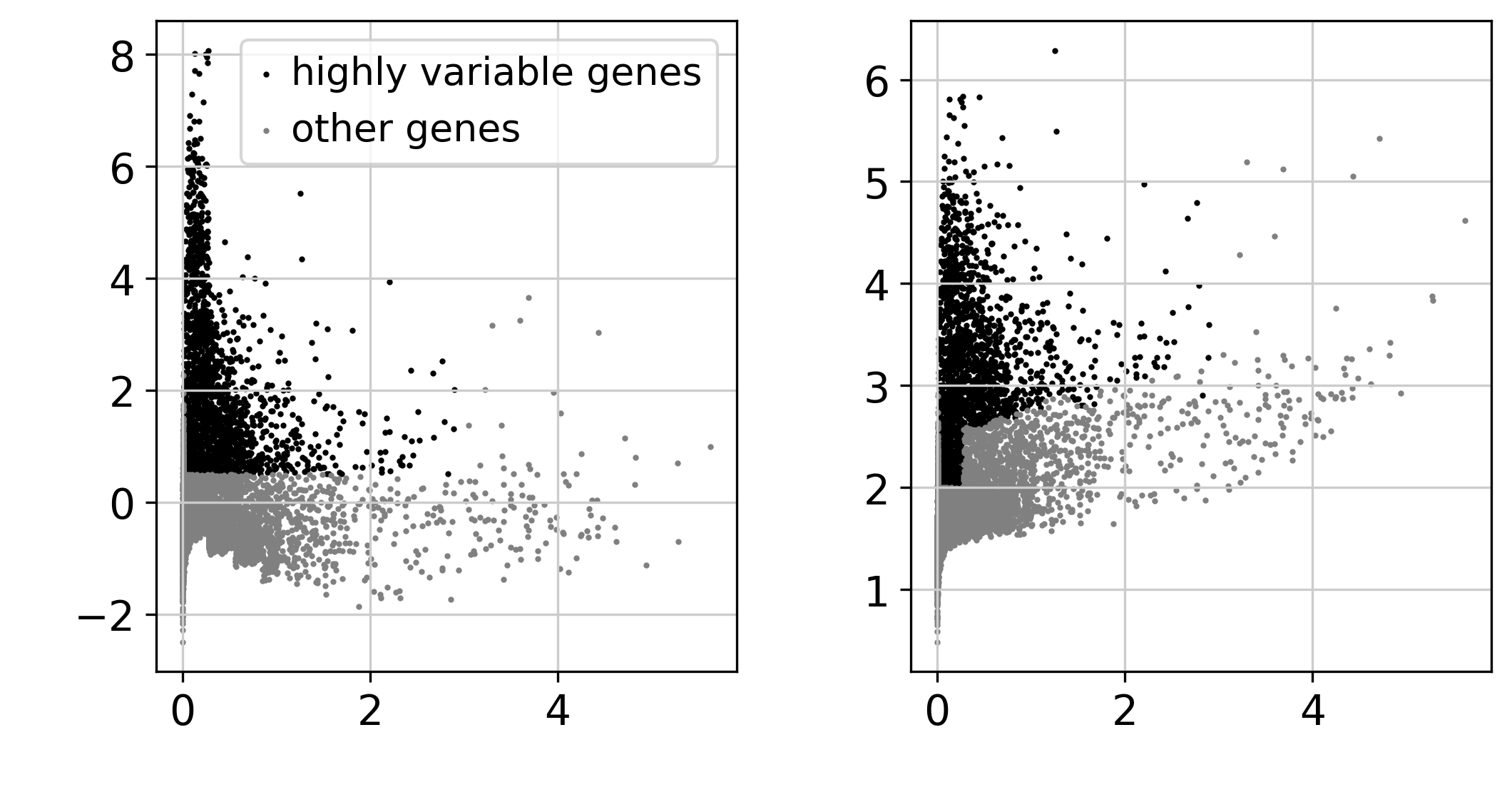
# 识别高变的基因
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
extracting highly variable genes
finished (0:00:00)
--> added
'highly_variable', boolean vector (adata.var)
'means', float vector (adata.var)
'dispersions', float vector (adata.var)
'dispersions_norm', float vector (adata.var)
sc.pl.highly_variable_genes(adata,
show=False)
plt.savefig("./fig2/highly_variable_plot.png", bbox_inches="tight", dpi=300)
## 过滤操作
adata.raw = adata.copy()
adata = adata[:, adata.var.highly_variable]
# 回归每个细胞的总计数和表达的线粒体基因百分比的影响。将数据缩放到单位方差。
sc.pp.regress_out(adata, ["total_counts", "pct_counts_mt"])
regressing out ['total_counts', 'pct_counts_mt']
sparse input is densified and may lead to high memory use
finished (0:00:07)
sc.pp.scale(adata, max_value=10)
## 保存为h5ad格式
adata.write('pbmc_scale.h5ad')