Scanpy单细胞处理流程之三:降维&注释
待补充: 怎么美化小提琴图 怎么绘制有细胞比例的umap图
#安装导入需要的模块
import numpy as np
import pandas as pd
import scanpy as sc
import os
import torch
import omicverse as ov
import matplotlib.pyplot as plt
from matplotlib import patheffects
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
Version: 1.6.10, Tutorials: https://omicverse.readthedocs.io/
# 检查当前的工作路径
current_working_directory = os.getcwd()
print(f"当前的工作路径: {current_working_directory}")
# 检查 torch 的 GPU 版本
if torch.cuda.is_available():
print(f"torch GPU 版本: {torch.version.cuda}")
else:
print("未检测到 GPU")
sc.settings.verbosity = 3# 日志详细程度
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80,facecolor='white')
当前的工作路径: /mnt/DEV_8T/zhaozm/scanpy_单细胞流程/pbmc_test
torch GPU 版本: 12.1
## 读取文件
adata = sc.read_h5ad('pbmc_scale.h5ad')
# 全局设置背景
plt.rcParams["figure.facecolor"] = "white"
plt.rcParams["savefig.facecolor"] = "white"
# 设置全局图例文字颜色
plt.rcParams["legend.labelcolor"] = "black"
plt.rcParams["text.color"] = "black"
## 检查是否进行了归一化
adata.X.max()
10.0
sc.tl.pca(adata, svd_solver="arpack")
computing PCA
with n_comps=50
finished (0:00:01)
## 查看基因的分布
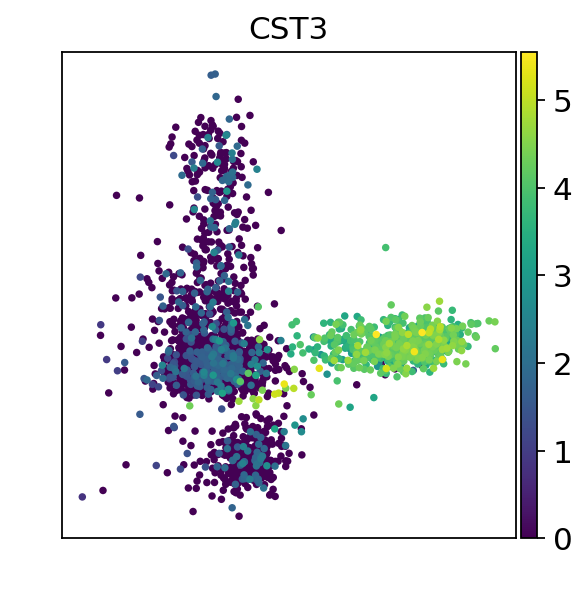
sc.pl.pca(adata, color="CST3",show=False)
plt.savefig("./fig3/gene_marker_plot.png", bbox_inches="tight", dpi=300)
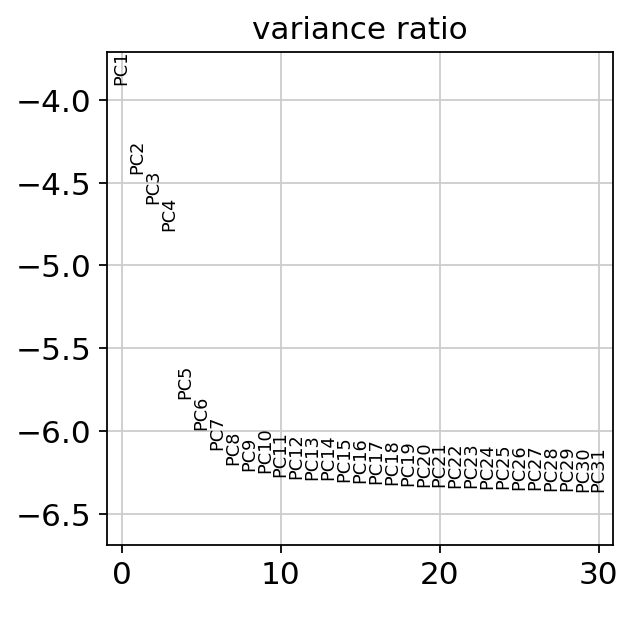
## 查看elbow
sc.pl.pca_variance_ratio(adata, log=True,show=False)
plt.savefig("./fig3/elbow_plot.png", bbox_inches="tight", dpi=300)
## 保存结果
adata.write('adata_pca.h5ad')
adata
AnnData object with n_obs × n_vars = 2638 × 1838
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'ribo', 'hb', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'hvg', 'log1p', 'pca'
obsm: 'X_pca'
varm: 'PCs'
# Computing the neighborhood graph
## 计算邻域图
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
## seurat里面n_neighbors的默认参数是20,由k.param 参数控制
computing neighbors
using 'X_pca' with n_pcs = 40
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:06)
## 聚类
sc.tl.leiden(
adata,
resolution=0.9,
random_state=0,
flavor="igraph",
n_iterations=2,
directed=False,
)
running Leiden clustering
finished: found 8 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:00)
## 绘制UMAP图
sc.tl.paga(adata)
sc.pl.paga(adata, plot=False) # remove `plot=False` if you want to see the coarse-grained graph
sc.tl.umap(adata, init_pos='paga')
running PAGA
finished: added
'paga/connectivities', connectivities adjacency (adata.uns)
'paga/connectivities_tree', connectivities subtree (adata.uns) (0:00:00)
--> added 'pos', the PAGA positions (adata.uns['paga'])
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm)
'umap', UMAP parameters (adata.uns) (0:00:04)
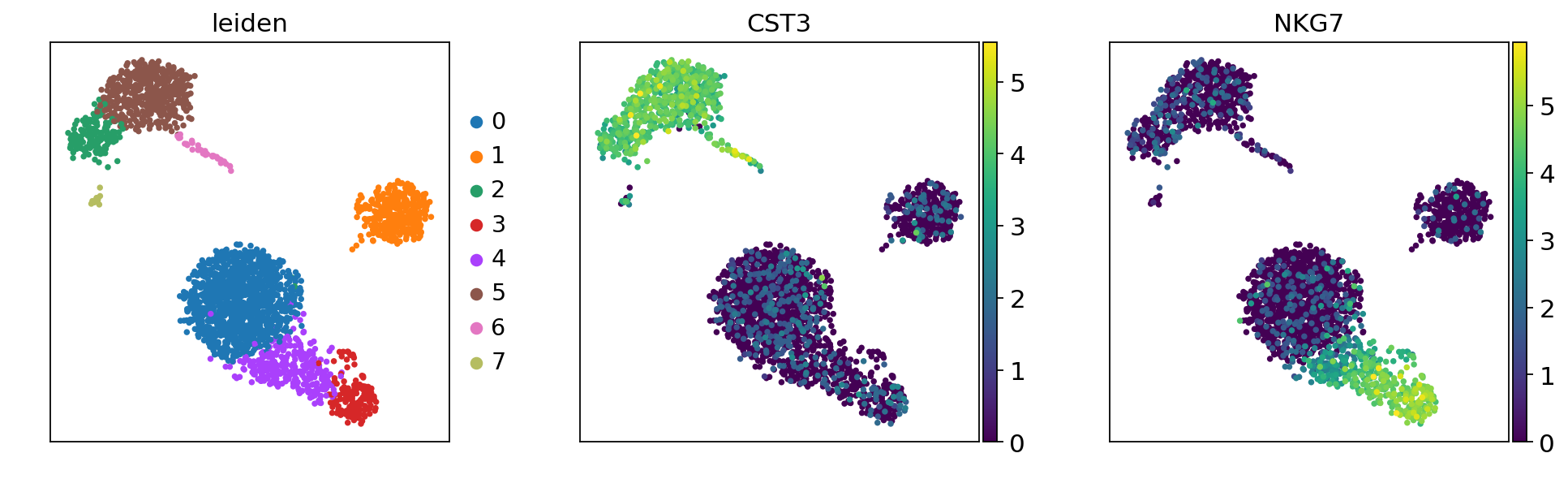
sc.pl.umap(adata, color=["leiden", "CST3", "NKG7"])
## 保存结果
adata.write('adata_umap.h5ad')
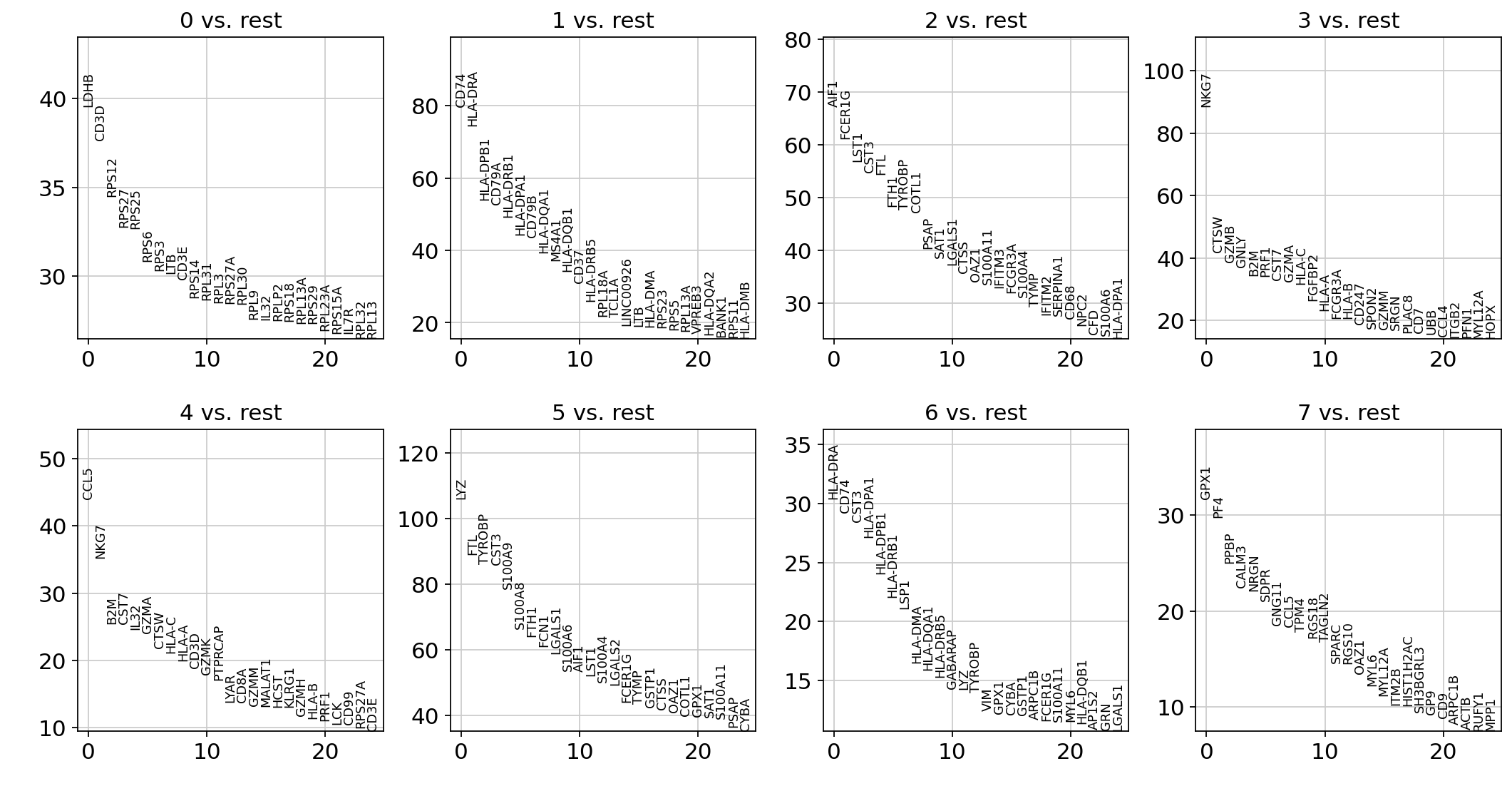
让我们来计算每个聚类中高差异基因的排名。 默认情况下,如果 AnnData 属性已经初始化,则会使用该属性。 最简单快捷的方法是 t 检验。但是R中使用的是wilcoxon秩和检验,审稿人也更认可wilcoxon秩和检验,在本次的数据集中,基因差距不是很大,但大规模的数据集里会产生差距。因此后面的计算使用wilcoxon秩和检验。
## 寻找高变基因
sc.tl.rank_genes_groups(adata, "leiden", method="t-test")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
ranking genes
finished: added to `.uns['rank_genes_groups']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:00)
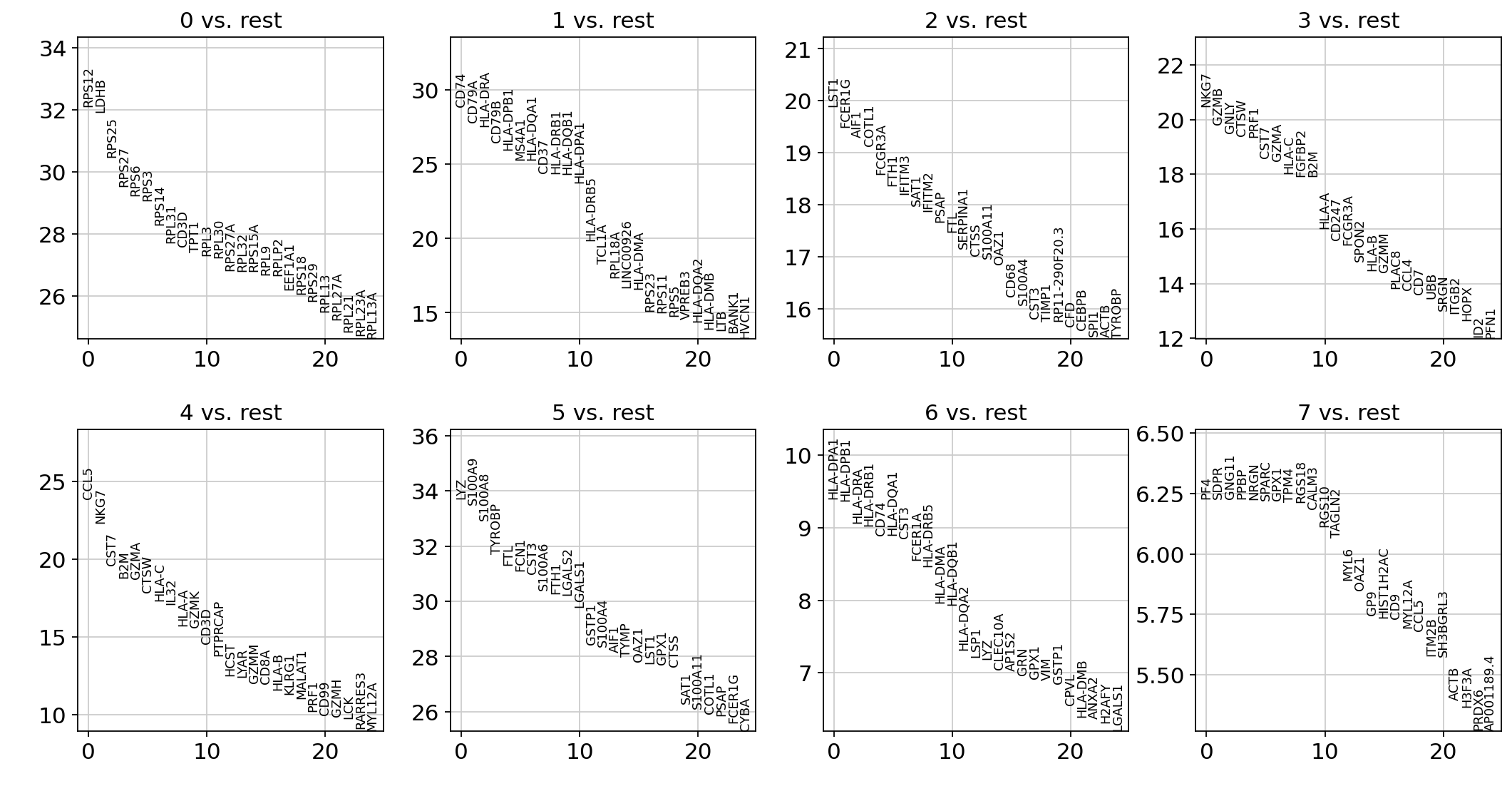
sc.tl.rank_genes_groups(adata, "leiden", method="wilcoxon")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
ranking genes
finished: added to `.uns['rank_genes_groups']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:02)
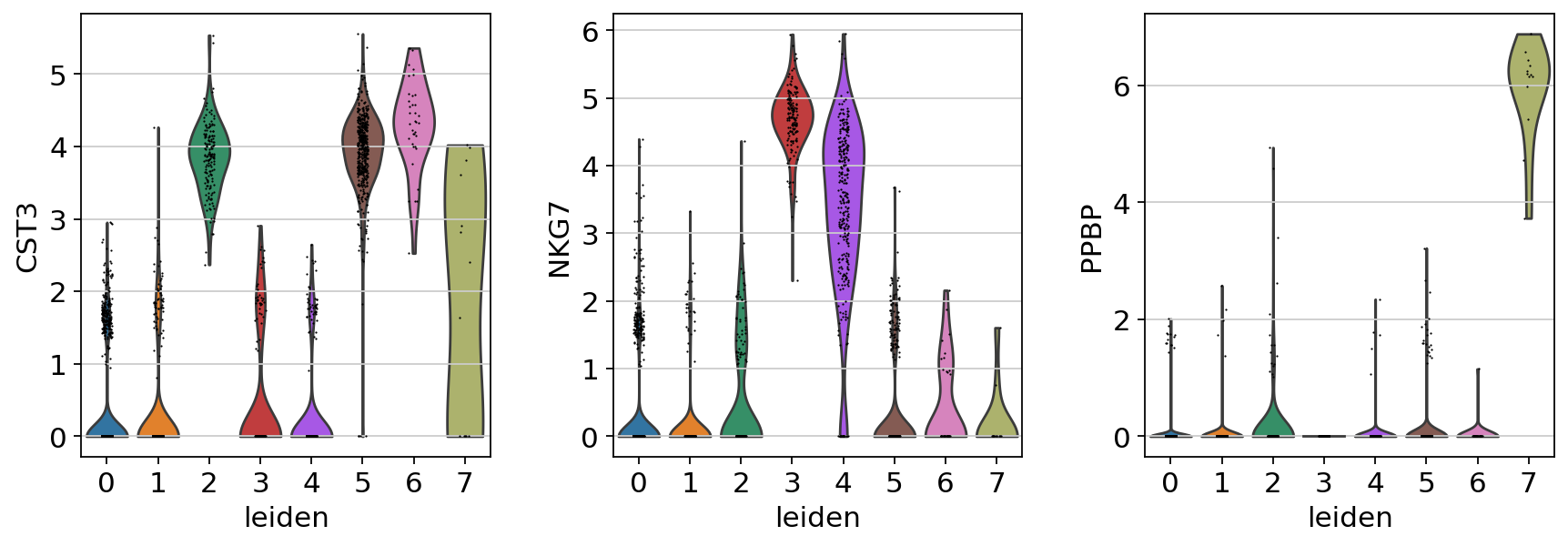
# 全局设置
plt.rcParams.update({
"axes.titlecolor": "black",
"text.color": "black",
"savefig.facecolor": "white",
"xtick.color": "black", # X轴刻度颜色
"ytick.color": "black", # Y轴刻度颜色
"axes.labelcolor": "black", # 坐标轴标签颜色
})
# 绘制小提琴图(不自动显示)
sc.pl.violin(
adata,
["CST3", "NKG7", "PPBP"],
groupby="leiden",
show=False
)
# 保存图像
plt.savefig("./fig3/viol_plot.png", bbox_inches="tight", dpi=300)
# 重命名 'leiden' 的分类标签
new_cluster_names = [
"CD4 T",
"B",
"FCGR3A+ Monocytes",
"NK",
"CD8 T",
"CD14+ Monocytes",
"Dendritic",
"Megakaryocytes",
]
adata.rename_categories("leiden", new_cluster_names)
# 将重命名后的分类复制到新列 'celltype'(保留原始数值标签)
adata.obs["celltype"] = adata.obs["leiden"].astype(str) # 直接复制分类后的字符串
adata
print(adata.obs['leiden'].head())
AAACATACAACCAC-1 CD4 T
AAACATTGAGCTAC-1 B
AAACATTGATCAGC-1 CD4 T
AAACCGTGCTTCCG-1 FCGR3A+ Monocytes
AAACCGTGTATGCG-1 NK
Name: leiden, dtype: category
Categories (8, object): ['CD4 T', 'B', 'FCGR3A+ Monocytes', 'NK', 'CD8 T', 'CD14+ Monocytes', 'Dendritic', 'Megakaryocytes']
下面使用omicverse包对所有的图片进行美化
## 小提琴图
## umap图1
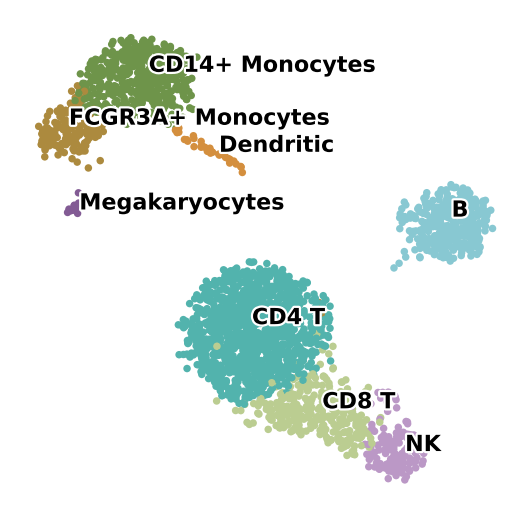
ov.utils.embedding(adata,
basis='X_umap',
color=[ "leiden"],
title=['Clusters'],
palette=ov.palette()[:],
show=False,frameon='small',)
<AxesSubplot: title={'center': 'Clusters'}, xlabel='X_umap1', ylabel='X_umap2'>

fig, ax = plt.subplots(figsize=(4,4))
celltypes = adata.obs["celltype"].cat.categories.tolist()
print("细胞类型顺序:", celltypes)
custom_palette = ov.pl.blue_color[4:6]+ov.pl.green_color[4:6]+ov.pl.orange_color[1:3]+ov.pl.purple_color[1:3]
# 创建颜色映射字典(celltype → color)
color_mapping = {ct: custom_palette[i] for i, ct in enumerate(celltypes)}
# 绘制 UMAP(按 celltype 着色)
ov.pl.embedding(
adata,
basis="X_umap",
color=['celltype'],
title='',
show=False,
legend_loc=None,
add_outline=False,
frameon='small',
legend_fontoutline=2,
ax=ax,
palette=color_mapping # 使用自定义颜色映射
)
# 添加聚类注释标签
ov.pl.embedding_adjust(
adata,
basis="X_umap",
groupby='leiden',
ax=ax,
adjust_kwargs=dict(arrowprops=dict(arrowstyle='-', color='black')),
text_kwargs=dict(
fontsize=10,
weight='bold',
color='black', # 标签文字颜色
path_effects=[patheffects.withStroke(linewidth=2, foreground='w')]
),
)
ax.axis("off")
fig.savefig('./fig3/umap-ct_pbmc.png', dpi=300, bbox_inches='tight')
细胞类型顺序: ['B', 'CD4 T', 'CD8 T', 'CD14+ Monocytes', 'Dendritic', 'FCGR3A+ Monocytes', 'Megakaryocytes', 'NK']