Seurat单细胞处理流程之一:数据读取
1.简介
单细胞数据格式主要划分为四大类:
- 10X标准文件(包含
barcodes.tsv.gz,features.tsv.gz和matrix.mtx.gz) - 表达矩阵文件(
csv,tsv,txt) - H5格式文件(
.h5) - H5AD格式文件(
.h5ad)
由于部分数据集包含多个样本但只提供了一个文件,因此数据读取的方式也变得相对复杂,四大类里面可划分为多个小类。
除了标准文件,其他格式对部分初学者来说都是一座大山。单细胞数据的读取,本质上就是:
- 表达量矩阵
- 行信息
- 列信息
- 构建Seurat对象
所谓知己知彼,百战不殆,理解数据是数据分析的前提。
2.10X标准文件的读取
10X标准文件包含cellranger上游比对分析产生的barcodes.tsv.gz,features.tsv.gz和matrix.mtx.gz 3个文件,分别代表细胞标签(barcode)、基因ID(feature)、表达数据(matrix) 。一般先使用read10X()函数对这三个文件进行整合,得到稀疏表达矩阵(行为基因、列为细胞,dgCMatrix格式)。如下图所示:
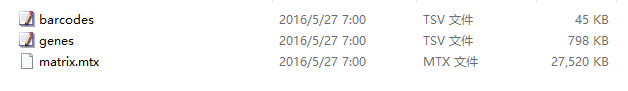
rm(list = ls())
pbmc.data <- Read10X(data.dir = "./filtered_gene_bc_matrices/hg19/")
#自动读取10X的数据,是一些tsv与mtx文件
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "biomamba")
这种读取方式比较简单,直接读取即可。
3.表达矩阵文件的读取
以数据集GSE179994为例,GEO官网的文件就是此种类型,其将表达量矩阵和行列信息分开存放了,对于此种数据,也应该分开读取并构造seurat对象。
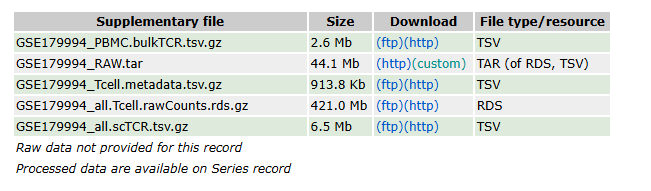
GSE179994_all.Tcell.rawCounts.rds这个文件存放的是表达矩阵信息,是一个稀疏矩阵。GSE179994_Tcell.metadata文件是行列信息,包含关于样本、细胞或实验设计的关键信息。
## 加载R包
library(Seurat)
library(dplyr)
library(readr)
library(Matrix)
library(ggplot2)
library(patchwork)
library(ggplot2)
set.seed(123)
# 读入数据
pbmc <- readRDS('./data/GSE179994_all.Tcell.rawCounts.rds')
pbmc
# 转换为数据框
pbmc <- as.data.frame(pbmc)
# 转置
pbmc <- as.data.frame(t(pbmc))
# 创建 seurant对象
pbmc <- CreateSeuratObject(counts = mergedata)
# 给 seurant对象添加元数据
metedata <- read.table("./data/GSE179994_Tcell.metadata.tsv",sep = "\t",header = T)
unique(metedata$sample)
# 提取metedata的列
patient <- metedata$sample
group <- metedata$Type
# 添加元数据
pbmc <- AddMetaData(object = pbmc, #seurat对象
metadata = patient, #需要添加的metadata
col.name = "sample") #给新添加的metadata命名
pbmc <- AddMetaData(object = pbmc,
metadata = group,
col.name = "group")
4.H5格式文件与H5AD格式文件的读取
h5格式是一种用于存储大规模数据的二进制文件格式,它可以包含多种数据类型,如矩阵、表格、图像等。在读取h5格式单细胞数据时,可使用Read10X_h5()函数创建Seurat对象。 示例数据集:GSE168215
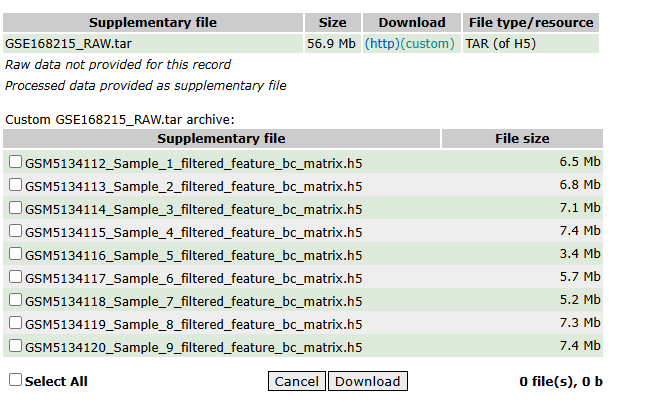
rm(list=ls())
options(stringsAsFactors = F)
library(dplyr)
library(Seurat)
library(data.table)
library(dplyr)
getwd()
dir='GSE168215_RAW/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[7]
print(pro)
tmp = Read10X_h5(file.path(dir,pro ))
if(length(tmp)==2){
ct = tmp[[1]]
}else{ct = tmp}
ct[1:10, 1:2]
tail( ct[ , 1:2])
rownames(ct)=gsub('GRCh38______','', rownames(ct))
sce =CreateSeuratObject(counts = ct ,
project = gsub('_filtered_feature_bc_matrix.h5','',pro) ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
5. H5AD格式文件的读取
h5ad格式是专门用于存储和分享单细胞表达数据,它使用Anndata库来创建和读取。h5ad格式可以与cellxgene或Seurat等工具兼容,进行单细胞数据的可视化和分析。但h5ad格式是python读取文件格式,如果你只会使用R语言,就需要先转换为seurat包可操作的对象。使用scanpy或其他工具可以完成转换。
rm(list=ls())
options(stringsAsFactors = F)
library(SeuratDisk)
library(patchwork)
getwd()
##h5ad是python的Scanpy读取文件格式,需要转换
Convert('./GSE153643_RAW/GSM4648564_adipose_raw_counts.h5ad', "h5seurat",
overwrite = TRUE,assay = "RNA")
scRNA <- LoadH5Seurat("./GSE153643_RAW/GSM4648564_adipose_raw_counts.h5seurat")
scRNA