Seurat单细胞处理流程之二:数据质控
1.简介
单细胞RNA测序(scRNA-seq)数据质量控制(Quality Control, QC)是分析流程中的关键步骤。由于单细胞测序技术存在一定的噪音和技术误差,质控的目的是去除低质量细胞和异常数据,提高后续分析的可靠性和生物学意义。
2.数据读入
以pbmc示例数据为例:
rm(list = ls())
setwd("/mnt/DEV_8T/zhaozm/seurat全流程/数据质控")
##禁止转化为因子
options(stringsAsFactors = FALSE)
library(Seurat)
library(dplyr)
library(readr)
library(Matrix)
library(ggplot2)
library(patchwork)
library(ggplot2)
载入需要的程序包:SeuratObject
载入需要的程序包:sp
载入程序包:‘SeuratObject’
The following objects are masked from ‘package:base’:
intersect, t
载入程序包:‘dplyr’
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
#读入数据
pbmc.data <- Read10X(data.dir = "filtered_gene_bc_matrices/hg19/")
## 创建seurat对象
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
Warning message:
“Feature names cannot have underscores ('_'), replacing with dashes ('-')”
#初步查看Seurat对象
pbmc
An object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
1 layer present: counts
3.质控数据及可视化
## 计算线粒体比例
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
#人源的数据为MT,鼠源的需要换成mt
head(pbmc@meta.data,5)
| orig.ident | nCount_RNA | nFeature_RNA | percent.mt | |
|---|---|---|---|---|
| <fct> | <dbl> | <int> | <dbl> | |
| AAACATACAACCAC-1 | pbmc3k | 2419 | 779 | 3.0177759 |
| AAACATTGAGCTAC-1 | pbmc3k | 4903 | 1352 | 3.7935958 |
| AAACATTGATCAGC-1 | pbmc3k | 3147 | 1129 | 0.8897363 |
| AAACCGTGCTTCCG-1 | pbmc3k | 2639 | 960 | 1.7430845 |
| AAACCGTGTATGCG-1 | pbmc3k | 980 | 521 | 1.2244898 |
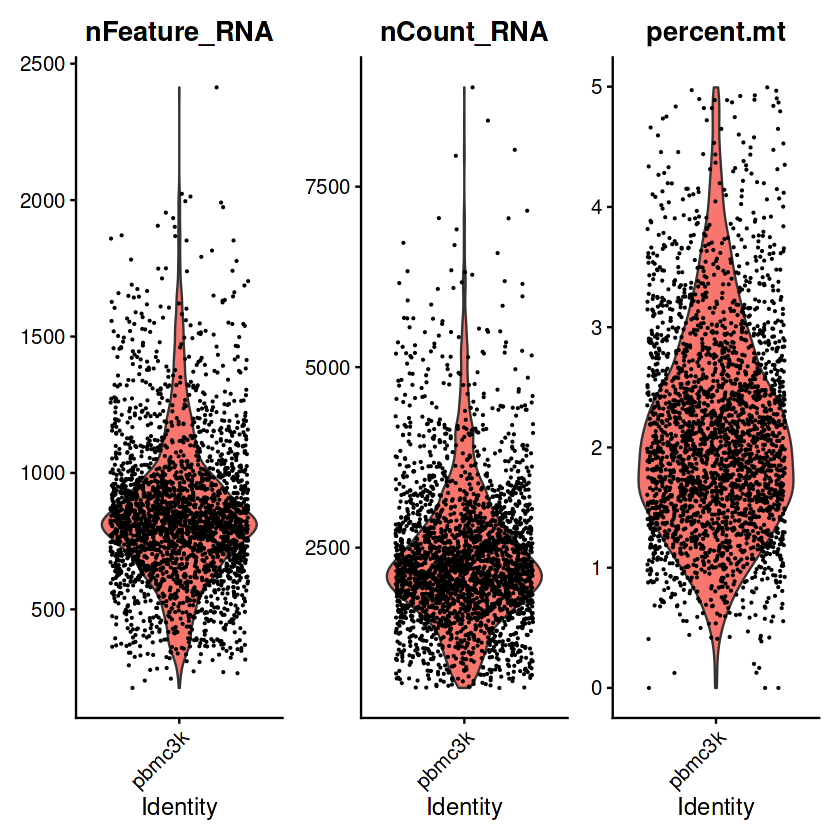
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
Warning message:
“Default search for "data" layer in "RNA" assay yielded no results; utilizing "counts" layer instead.”
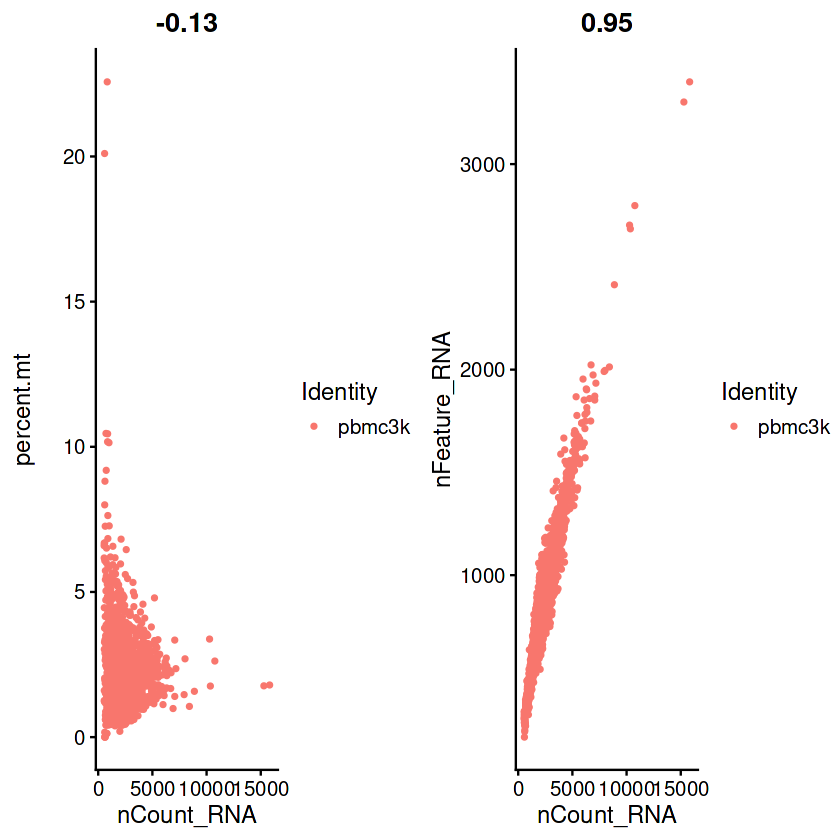
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
if(!require(patchwork))install.packages("patchwork")
CombinePlots(plots = list(plot1, plot2))
Warning message in CombinePlots(plots = list(plot1, plot2)):
“CombinePlots is being deprecated. Plots should now be combined using the patchwork system.”
## 过滤
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
ncol(as.data.frame(GetAssayData(pbmc, slot = "counts")))
## [1] 2638
Warning message:
“[1m[22mThe `slot` argument of `GetAssayData()` is deprecated as of SeuratObject 5.0.0.
[36mℹ[39m Please use the `layer` argument instead.”
2638
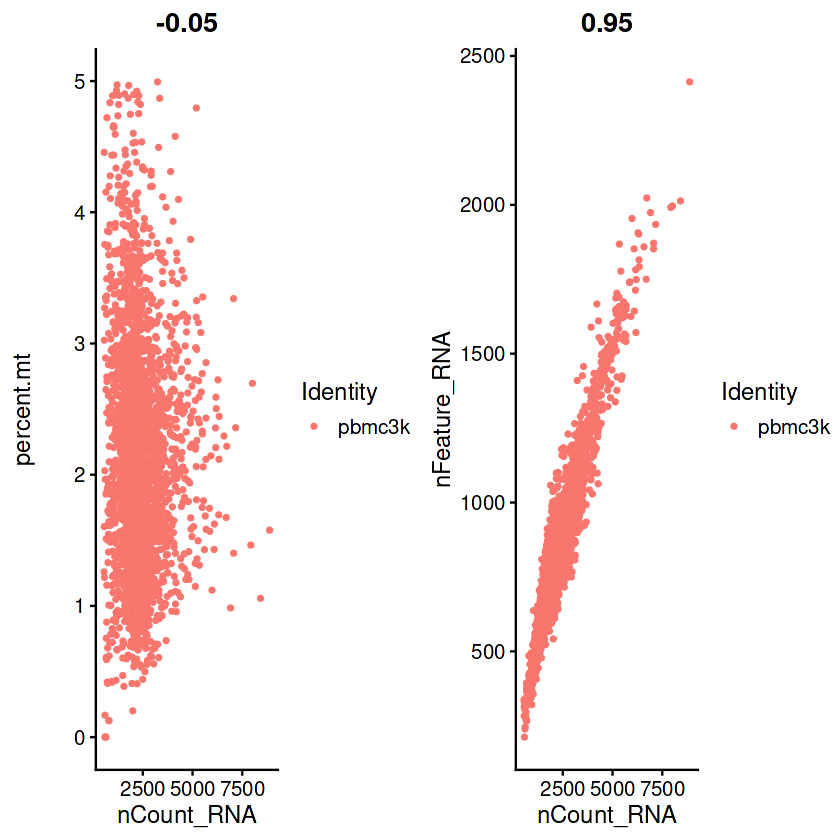
## 过滤之后的图片
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
if(!require(patchwork))install.packages("patchwork")
#CombinePlots这步需要你的绘图窗口足够大
CombinePlots(plots = list(plot1, plot2))
Warning message in CombinePlots(plots = list(plot1, plot2)):
“CombinePlots is being deprecated. Plots should now be combined using the patchwork system.”
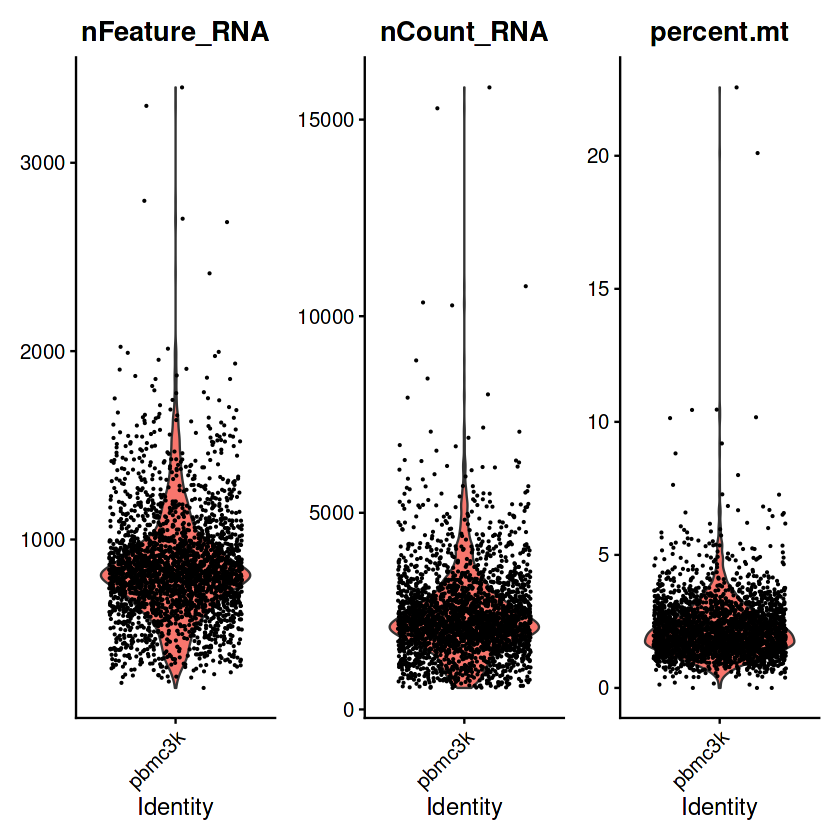
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
Warning message:
“Default search for "data" layer in "RNA" assay yielded no results; utilizing "counts" layer instead.”