Seurat单细胞处理流程之四:差异分析&富集分析
简介
整体来说,单细胞的差异分析与普通转录组的差异分析流程类似,但是原理略有不同。 (补充几个常用包的原理和秩和检验的原理)
1.差异分析
rm(list = ls())
setwd("/mnt/DEV_8T/zhaozm/seurat全流程/差异富集/")
##禁止转化为因子
options(stringsAsFactors = FALSE)
## 设置保存的目录
## 数据目录
data_dir <- "/mnt/DEV_8T/zhaozm/seurat全流程/差异富集/data"
if (!dir.exists(data_dir)) {
dir.create(data_dir, recursive = TRUE)
}
## 图片目录
img_dir <- "/mnt/DEV_8T/zhaozm/seurat全流程/差异富集/img"
if (!dir.exists(img_dir)) {
dir.create(img_dir, recursive = TRUE)
}
library(readr)
library(gplots)
library(ggplot2)
library(Seurat)
library(scRNAtoolVis)
library(dplyr)
载入需要的程序包:tidyverse
## 读取注释之后的文件
pbmc <- read_rds(file = "./data/cd8注释后.rds")
## 删除所有的线粒体基因和核糖体基因后再进行差异分析
exp_m <- GetAssayData(pbmc, layer = 'counts')
gene.all <- rownames(exp_m)
mt.gene = grep('^MT-', gene.all, value = TRUE)#线粒体基因
ribosome.gene = grep('^RPL|^RPS|^MRPS|^MRPL', gene.all, value = TRUE)#核糖体基因
features = setdiff(gene.all, c(mt.gene, ribosome.gene))#去除不需要的基因
unique(pbmc$group)
- Resistant
- Pre
- Sensitive
# 差异表达分析函数
perform_deg_analysis <- function(cell_subset, ident1, ident2) {
FindMarkers(
object = cell_subset,
logfc.threshold = 0.25,
min.pct = 0.1,
only.pos = FALSE,
features = features,
ident.1 = ident1,
ident.2 = ident2
) %>% mutate(gene = rownames(.))
}
## 阈值可以修改
# 筛选函数
filter_degs <- function(deg_data) {
deg_data %>%
filter(pct.1 > 0.1 & p_val < 0.05) %>%
filter(abs(avg_log2FC) > 0.5)
}
## 提取tex—cd8
tex_cd8 <- subset(pbmc, subset = celltype == "Tex_CD8")
## 执行差异分析
Idents(tex_cd8)="group"
tex_cd8_degs <- perform_deg_analysis(tex_cd8, "Resistant", "Sensitive")
tex_cd8_degs_fil <- filter_degs(tex_cd8_degs)
## 保存ex的差异基因
write.csv(tex_cd8_degs_fil,file = "./data/tex_cd8_差异基因.csv")
# 全部细胞的差异分析 ---------------------------------------------------------------
Idents(pbmc) <- pbmc$group
# 定义一个函数来处理每个细胞类型
process_cell_type <- function(cell_type, pbmc) {
# 子集化为特定细胞类型
cell_subset <- subset(pbmc, subset = celltype == cell_type)
# 查找差异表达基因
degs <- FindMarkers(cell_subset,
logfc.threshold = 0.25,
min.pct = 0.1,
only.pos = FALSE,
features = features,
ident.1 = "Resistant", ident.2 = "Sensitive") %>%
mutate(gene = rownames(.))
# 过滤符合条件的差异表达基因
degs_filtered <- degs %>%
filter(pct.1 > 0.1 & p_val_adj < 0.05) %>%
filter(abs(avg_log2FC) > 0.5)
# 返回过滤后的结果,并标记是哪个细胞类型
return(list(cell_type = cell_type, degs_filtered = degs_filtered))
}
# 获取所有独特的细胞类型
cell_types <- unique(pbmc$celltype)
cell_types
# 使用 lapply 对每个细胞类型进行处理
results <- lapply(cell_types, function(cell_type) process_cell_type(cell_type, pbmc))
- Trm_CD8
- Cytotox_CD8
- Naive_CD8
- Tem_CD8
- Tex_CD8
# 将结果组合成一个数据框
library(dplyr)
# 组合所有细胞类型的差异表达基因数据
combined_results <- do.call(rbind, lapply(results, function(x) {
x$degs_filtered %>% mutate(cell_type = x$cell_type)
}))
# 将最后一列改成cluster
colnames(combined_results)[colnames(combined_results) == "cell_type"] <- "cluster"
combined_results$cluster <- as.factor(combined_results$cluster)
## 保存差异结果
head(combined_results)
table(combined_results$cluster)
write.csv(combined_results,file = "./data/cd8差异结果.csv")
combined_results <- read.csv(file = "./data/cd8差异结果.csv")
| p_val | avg_log2FC | pct.1 | pct.2 | p_val_adj | gene | cluster | |
|---|---|---|---|---|---|---|---|
| <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <chr> | <fct> | |
| HLA-B | 1.884343e-244 | 0.6819049 | 0.999 | 0.976 | 3.729116e-240 | HLA-B | Trm_CD8 |
| PCBP2 | 5.772154e-190 | 1.1862094 | 0.736 | 0.454 | 1.142309e-185 | PCBP2 | Trm_CD8 |
| ZFP36 | 4.087883e-127 | 0.7755835 | 0.912 | 0.764 | 8.089920e-123 | ZFP36 | Trm_CD8 |
| CCL4L2 | 4.095107e-113 | 1.8297831 | 0.676 | 0.447 | 8.104217e-109 | CCL4L2 | Trm_CD8 |
| RBM38 | 2.756488e-83 | 1.0418639 | 0.517 | 0.311 | 5.455089e-79 | RBM38 | Trm_CD8 |
| ZFP36L2 | 4.115279e-80 | 0.5919972 | 0.926 | 0.827 | 8.144138e-76 | ZFP36L2 | Trm_CD8 |
Cytotox_CD8 Naive_CD8 Tem_CD8 Tex_CD8 Trm_CD8
100 5 34 346 143
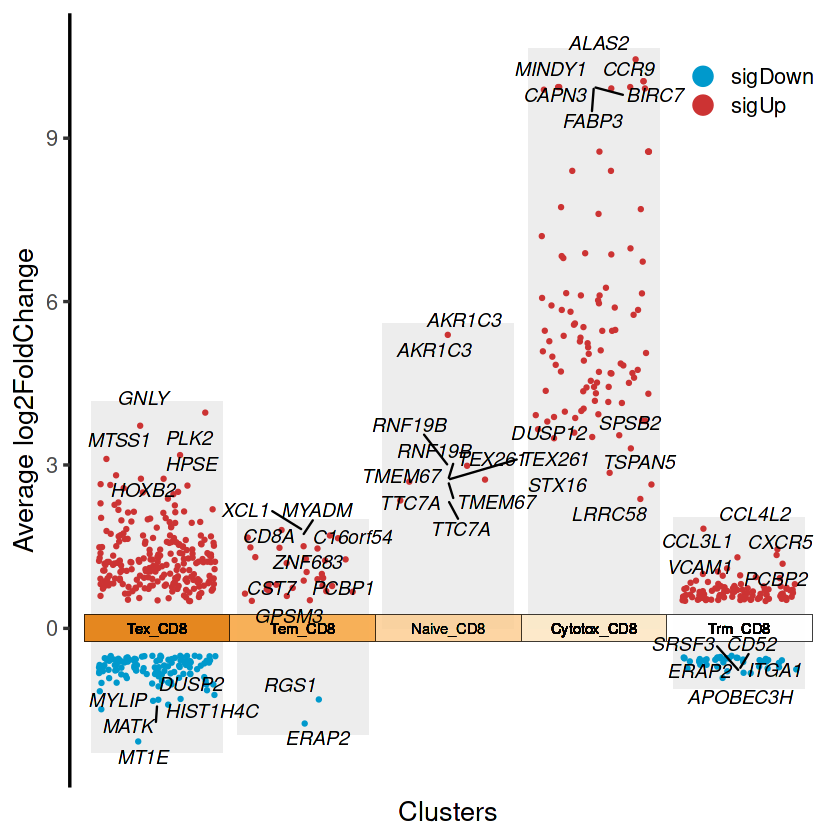
jjVolcano(diffData = combined_results,
tile.col = corrplot::COL2('PuOr', 15)[4:12],
size = 4,
fontface = 'italic',
base_size = 16,
legend.position = c(0.9, 0.9),
cluster.order = rev(unique(combined_results$cluster)))
2.富集分析
rm(list = ls())
setwd("/mnt/DEV_8T/zhaozm/seurat全流程/差异富集/")
##禁止转化为因子
options(stringsAsFactors = FALSE)
## 设置保存的目录
## 数据目录
data_dir <- "/mnt/DEV_8T/zhaozm/seurat全流程/差异富集/data"
if (!dir.exists(data_dir)) {
dir.create(data_dir, recursive = TRUE)
}
## 图片目录
img_dir <- "/mnt/DEV_8T/zhaozm/seurat全流程/差异富集/img"
if (!dir.exists(img_dir)) {
dir.create(img_dir, recursive = TRUE)
}
## 加载R包
library(readr)
library(msigdbr)
library(gplots)
library(ggplot2)
library(clusterProfiler)
library(org.Hs.eg.db)
library(Seurat)
library(singleseqgset)
library(scRNAtoolVis)
library(dplyr)
载入程序包:‘gplots’
The following object is masked from ‘package:stats’:
lowess
clusterProfiler v4.14.4 Learn more at https://yulab-smu.top/contribution-knowledge-mining/
Please cite:
Guangchuang Yu, Li-Gen Wang, Yanyan Han and Qing-Yu He.
clusterProfiler: an R package for comparing biological themes among
gene clusters. OMICS: A Journal of Integrative Biology. 2012,
16(5):284-287
载入程序包:‘clusterProfiler’
The following object is masked from ‘package:stats’:
filter
载入需要的程序包:AnnotationDbi
载入需要的程序包:stats4
载入需要的程序包:BiocGenerics
载入程序包:‘BiocGenerics’
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, aperm, append, as.data.frame, basename, cbind,
colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
Position, rank, rbind, Reduce, rownames, sapply, saveRDS, setdiff,
table, tapply, union, unique, unsplit, which.max, which.min
载入需要的程序包:Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
载入需要的程序包:IRanges
载入需要的程序包:S4Vectors
载入程序包:‘S4Vectors’
The following object is masked from ‘package:clusterProfiler’:
rename
The following object is masked from ‘package:gplots’:
space
The following object is masked from ‘package:utils’:
findMatches
The following objects are masked from ‘package:base’:
expand.grid, I, unname
载入程序包:‘IRanges’
The following object is masked from ‘package:clusterProfiler’:
slice
载入程序包:‘AnnotationDbi’
The following object is masked from ‘package:clusterProfiler’:
select
载入需要的程序包:SeuratObject
载入需要的程序包:sp
载入程序包:‘sp’
The following object is masked from ‘package:IRanges’:
%over%
载入程序包:‘SeuratObject’
The following object is masked from ‘package:IRanges’:
intersect
The following object is masked from ‘package:S4Vectors’:
intersect
The following object is masked from ‘package:BiocGenerics’:
intersect
The following objects are masked from ‘package:base’:
intersect, t
载入需要的程序包:Matrix
载入程序包:‘Matrix’
The following object is masked from ‘package:S4Vectors’:
expand
载入需要的程序包:tidyverse
## 富集分析需要挂上代理
Sys.setenv("http_proxy"="http://10.16.46.126:7890")
Sys.setenv("https_proxy"="http://10.16.46.126:7890")
## 读取差异分析之后的结果
tex_cd8_degs_fil <- read.csv(file = "./data/tex_cd8_差异基因.csv")
head(tex_cd8_degs_fil)
| X | p_val | avg_log2FC | pct.1 | pct.2 | p_val_adj | gene | |
|---|---|---|---|---|---|---|---|
| <chr> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <chr> | |
| 1 | GNLY | 3.132400e-85 | 3.961162 | 0.543 | 0.147 | 6.199019e-81 | GNLY |
| 2 | DUSP2 | 8.528781e-80 | -1.330854 | 0.772 | 0.969 | 1.687846e-75 | DUSP2 |
| 3 | ENTPD1 | 2.926838e-58 | 2.506306 | 0.353 | 0.069 | 5.792213e-54 | ENTPD1 |
| 4 | KLRD1 | 6.800869e-58 | 2.146381 | 0.476 | 0.139 | 1.345892e-53 | KLRD1 |
| 5 | CALM1 | 5.550144e-57 | -0.838229 | 0.868 | 0.975 | 1.098373e-52 | CALM1 |
| 6 | GAPDH | 7.037207e-56 | 1.164210 | 0.969 | 0.978 | 1.392663e-51 | GAPDH |
ids_tex <- bitr(tex_cd8_degs_fil$gene, 'SYMBOL', 'ENTREZID', OrgDb = org.Hs.eg.db)
head(ids_tex)
'select()' returned 1:1 mapping between keys and columns
Warning message in bitr(tex_cd8_degs_fil$gene, "SYMBOL", "ENTREZID", OrgDb = org.Hs.eg.db):
“2.64% of input gene IDs are fail to map...”
| SYMBOL | ENTREZID | |
|---|---|---|
| <chr> | <chr> | |
| 1 | GNLY | 10578 |
| 2 | DUSP2 | 1844 |
| 3 | ENTPD1 | 953 |
| 4 | KLRD1 | 3824 |
| 5 | CALM1 | 801 |
| 6 | GAPDH | 2597 |
tex_cd8_degs_fil <- merge(tex_cd8_degs_fil, ids_tex, by.x = 'gene', by.y = 'SYMBOL')
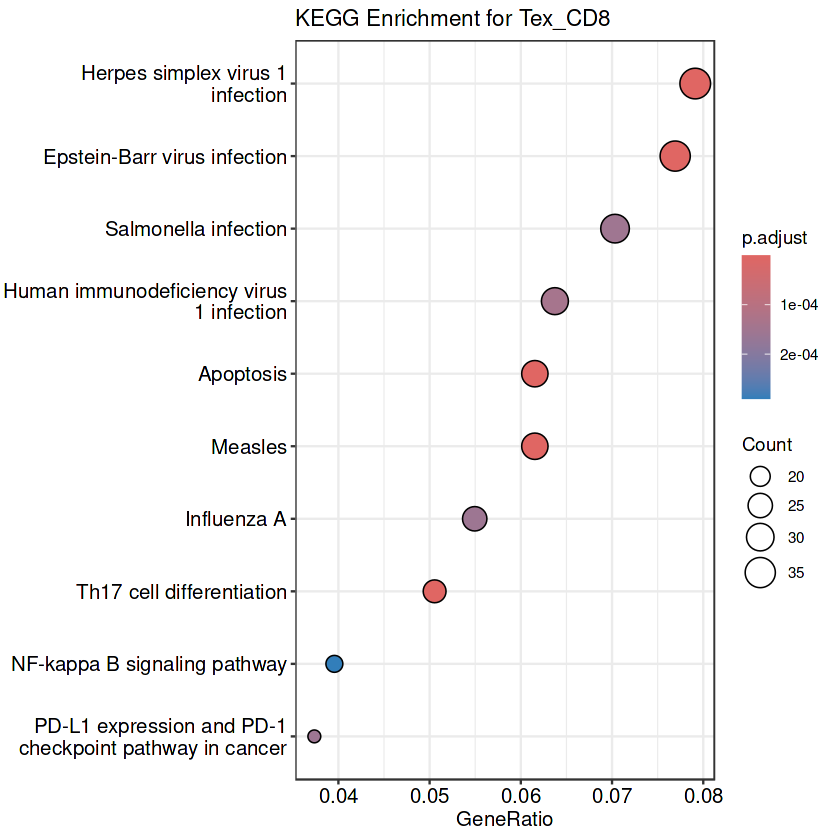
tex_cd8_kegg <- enrichKEGG(gene = tex_cd8_degs_fil$ENTREZID.x, organism = "hsa", pvalueCutoff = 1)
write.csv(as.data.frame(tex_cd8_kegg@result), file = "./data/tex_cd8_kegg_result.csv", row.names = FALSE)
dotplot(tex_cd8_kegg, showCategory = 10, title = "KEGG Enrichment for Tex_CD8")
## GSEA
library(clusterProfiler)
library(org.Hs.eg.db)
library(dplyr)
library(clusterProfiler)
library(enrichplot)
library(ggupset)
library(cowplot)
enrichplot v1.26.5 Learn more at https://yulab-smu.top/contribution-knowledge-mining/
Please cite:
Guangchuang Yu, Fei Li, Yide Qin, Xiaochen Bo, Yibo Wu and Shengqi
Wang. GOSemSim: an R package for measuring semantic similarity among GO
terms and gene products. Bioinformatics. 2010, 26(7):976-978
载入程序包:‘cowplot’
The following object is masked from ‘package:lubridate’:
stamp
tex_cd8_degs_fil <- tex_cd8_degs_fil[order(tex_cd8_degs_fil$avg_log2FC, decreasing = TRUE),]
tex_markers_list <- setNames(as.numeric(tex_cd8_degs_fil$avg_log2FC), tex_cd8_degs_fil$ENTREZID.x)
tex_cd8_gsea_go <- gseGO(geneList = tex_markers_list, OrgDb = org.Hs.eg.db, ont = "ALL", pvalueCutoff = 0.05)
tex_cd8_gsea_go_arrange <- arrange(as.data.frame(tex_cd8_gsea_go@result), desc(abs(NES)))
write.csv(tex_cd8_gsea_go_arrange, file = "./data/tex_cd8_gsea_go_results.csv", row.names = FALSE)
using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
preparing geneSet collections...
GSEA analysis...
Warning message in fgseaMultilevel(pathways = pathways, stats = stats, minSize = minSize, :
“There were 2 pathways for which P-values were not calculated properly due to unbalanced (positive and negative) gene-level statistic values. For such pathways pval, padj, NES, log2err are set to NA. You can try to increase the value of the argument nPermSimple (for example set it nPermSimple = 10000)”
Warning message in fgseaMultilevel(pathways = pathways, stats = stats, minSize = minSize, :
“For some of the pathways the P-values were likely overestimated. For such pathways log2err is set to NA.”
leading edge analysis...
done...
# 定义配色
color <- c("#f7ca64", "#43a5bf", "#86c697", "#a670d6", "#ef998a")
# 绘制 tex_cd8 的 GSEA-GO 图
## 上调
# 提取上调通路
upregulated_pathways <- tex_cd8_gsea_go@result %>%
filter(NES > 0) %>% # 筛选上调通路
arrange(desc(NES)) %>% # 按NES降序排列
slice(1:5) # 选择前5条通路
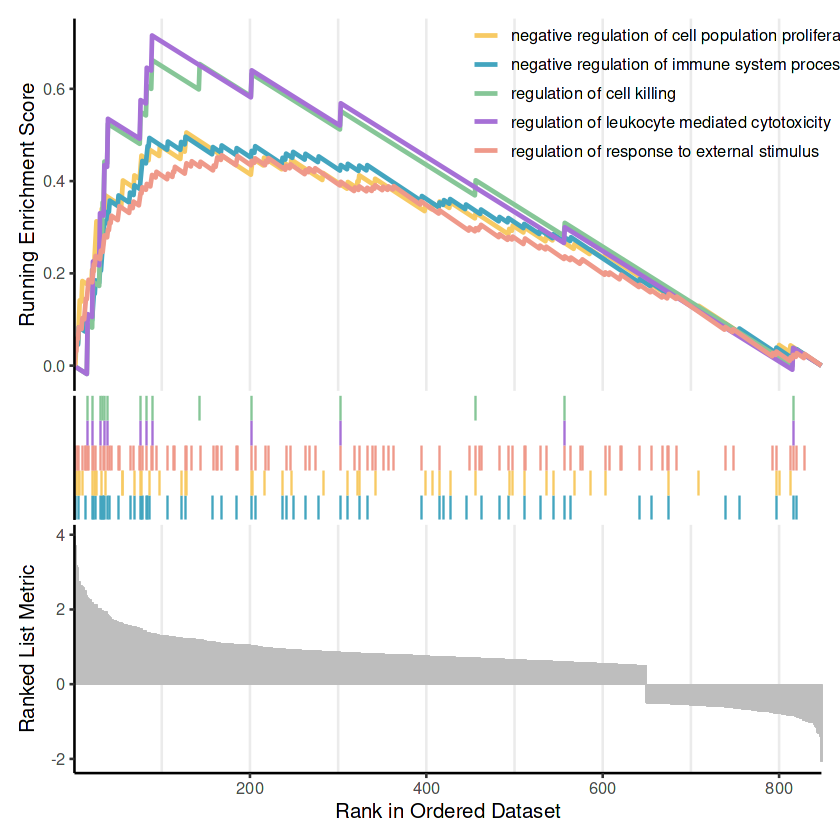
# 绘制前5条上调通路的GSEA曲线
gsekp1_tex <- gseaplot2(
tex_cd8_gsea_go, # GSEA结果对象
geneSetID = upregulated_pathways$ID, # 上调通路的ID向量
pvalue_table = F, # 是否显示p值表
base_size = 12, # 字体大小
color = color # 可选颜色
)
upregulated_pathways$Description
# 显示图形
gsekp1_tex
- negative regulation of immune system process
- regulation of response to external stimulus
- negative regulation of cell population proliferation
- regulation of leukocyte mediated cytotoxicity
- regulation of cell killing