载入需要的程序包:SeuratObject
载入需要的程序包:sp
载入程序包:‘SeuratObject’
The following objects are masked from ‘package:base’:
intersect, t
Using cached data manifest, last updated at 2025-03-11 16:14:29.676313
── �[1mInstalled datasets�[22m ──────────────────────────────── SeuratData v0.2.2.9001 ──
�[32m✔�[39m �[34mpbmc3k�[39m 3.1.4
────────────────────────────────────── Key ─────────────────────────────────────
�[32m✔�[39m Dataset loaded successfully
�[33m❯�[39m Dataset built with a newer version of Seurat than installed
�[31m❓�[39m Unknown version of Seurat installed
载入需要的程序包:foreach
载入需要的程序包:iterators
载入需要的程序包:parallel
## 执行gsva分析
#### Seurat V5对象 ####
## HALLMARKE基因集
ssc_dataset1 <- irGSEA.score(object = pbmc, assay = "RNA",
slot = "data", seeds = 123,
ncores = 8,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = "Homo sapiens",
category = "H",
subcategory = NULL,
geneid = "symbol",
method = c("AUCell","UCell","singscore",
"ssgsea", "JASMINE", "viper"),
aucell.MaxRank = NULL,
ucell.MaxRank = NULL,
kcdf = 'Gaussian')
Validating object structure
Updating object slots
Ensuring keys are in the proper structure
Updating matrix keys for DimReduc ‘pca’
Updating matrix keys for DimReduc ‘umap’
Ensuring keys are in the proper structure
Ensuring feature names don't have underscores or pipes
Updating slots in RNA
Updating slots in RNA_nn
Setting default assay of RNA_nn to RNA
Updating slots in RNA_snn
Setting default assay of RNA_snn to RNA
Updating slots in pca
Updating slots in umap
Setting umap DimReduc to global
Setting assay used for NormalizeData.RNA to RNA
Setting assay used for FindVariableFeatures.RNA to RNA
Setting assay used for ScaleData.RNA to RNA
Setting assay used for RunPCA.RNA to RNA
Setting assay used for FindNeighbors.RNA.pca to RNA
No assay information could be found for FindClusters
Setting assay used for RunUMAP.RNA.pca to RNA
Validating object structure for Assay5 ‘RNA’
Validating object structure for Graph ‘RNA_nn’
Validating object structure for Graph ‘RNA_snn’
Validating object structure for DimReduc ‘pca’
Validating object structure for DimReduc ‘umap’
Object representation is consistent with the most current Seurat version
Calculate AUCell scores
Finish calculate AUCell scores
Calculate UCell scores
Finish calculate UCell scores
Calculate singscore scores
Finish calculate singscore scores
Calculate ssgsea scores
�[36mℹ�[39m GSVA version 2.0.4
�[33m!�[39m 1 genes with constant values throughout the samples
�[33m!�[39m 7 genes with constant non-zero values throughout the samples
�[36mℹ�[39m Using a MulticoreParam parallel back-end with 8 workers
�[36mℹ�[39m Calculating ssGSEA scores for 50 gene sets
�[36mℹ�[39m Calculating ranks
�[36mℹ�[39m Calculating rank weights
�[32m✔�[39m Calculations finished
Finish calculate ssgsea scores
Calculate JASMINE scores
Finish calculate jasmine scores
Calculate viper scores
Finish calculate viper scores
# 整合差异基因集
# 如果报错,考虑加句代码options(future.globals.maxSize = 100000 * 1024^5)
result.dge1 <- irGSEA.integrate(object = ssc_dataset1,
group.by = "celltype",method = c("AUCell","UCell","singscore",
"ssgsea", "JASMINE", "viper"))
Calculate differential gene set : AUCell
Finish!
Calculate differential gene set : UCell
Finish!
Calculate differential gene set : singscore
Finish!
Calculate differential gene set : ssgsea
Finish!
Calculate differential gene set : JASMINE
Finish!
Calculate differential gene set : viper
Finish!
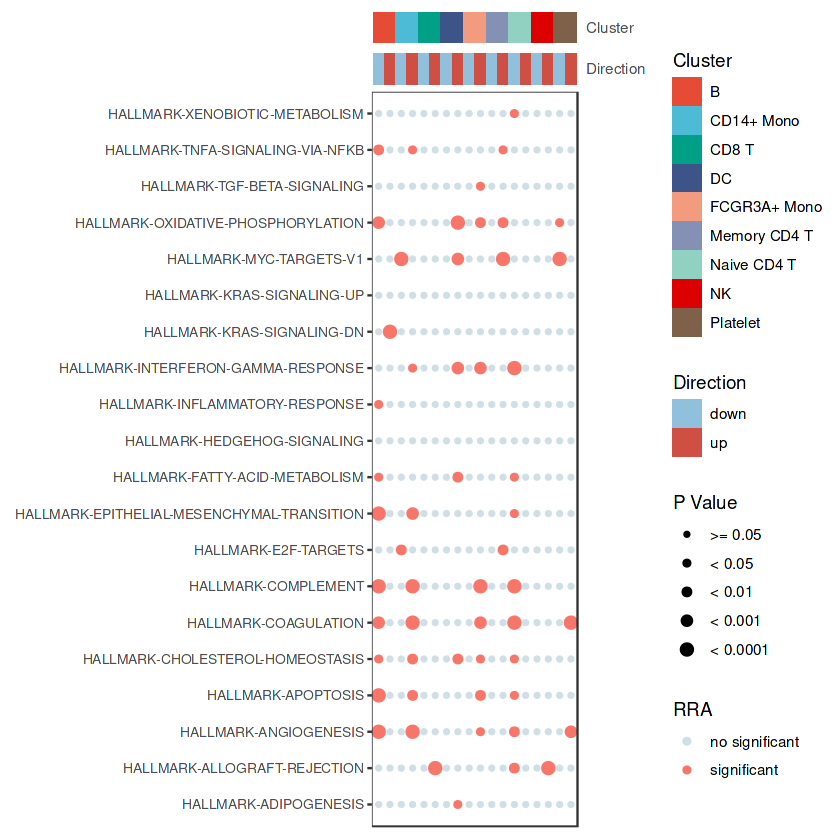
## 气泡图
bubble.plot1 <- irGSEA.bubble(object = result.dge1,
method = "RRA",
top = 20,
show.geneset = NULL,
cluster.color = pal_npg()(10),
direction.color = c('#90C0DC','#CF4F45'),
cluster_rows = F)
bubble.plot1
## GO 的富集类似,受运行时间所限,此处不再展示
pbmc3k.final <- irGSEA.score(object = pbmc,assay = "RNA",
slot = "data", seeds = 123, ncores = 8,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = "Homo sapiens", category = "C5",
subcategory = "GO:BP", geneid = "symbol",
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE"),
aucell.MaxRank = NULL, ucell.MaxRank = NULL,
kcdf = 'Gaussian')