Seurat单细胞处理流程之八:pySCENIC转录因子分析-R可视化
rm(list = ls())
setwd("/mnt/DEV_8T/zhaozm/seurat全流程/pyscenic/")
##禁止转化为因子
options(stringsAsFactors = FALSE)
## 设置保存的目录
## 数据目录
data_dir <- "/mnt/DEV_8T/zhaozm/seurat全流程/pyscenic/data"
if (!dir.exists(data_dir)) {
dir.create(data_dir, recursive = TRUE)
}
## 图片目录
img_dir <- "/mnt/DEV_8T/zhaozm/seurat全流程/pyscenic/img"
if (!dir.exists(img_dir)) {
dir.create(img_dir, recursive = TRUE)
}
options(repr.plot.width = 16, repr.plot.height = 10)
## 设置jupyter画板的全局参数
##可视化
library(Seurat)
library(SCopeLoomR)
library(AUCell)
library(SCENIC)
library(dplyr)
library(KernSmooth)
library(RColorBrewer)
library(plotly)
library(BiocParallel)
library(grid)
library(ComplexHeatmap)
library(data.table)
library(scRNAseq)
library(patchwork)
library(ggplot2)
library(stringr)
library(circlize)
## 读取上一步的loom文件
loom <- open_loom('./out_pbmc_SCENIC.loom')
regulons_incidMat <- get_regulons(loom, column.attr.name="Regulons")
regulons_incidMat[1:4,1:4]
| AL627309.1 | AP006222.2 | RP11-206L10.2 | RP11-206L10.9 | |
|---|---|---|---|---|
| AHR(+) | 0 | 0 | 0 | 0 |
| APEX1(+) | 0 | 0 | 0 | 0 |
| ARID3A(+) | 0 | 0 | 0 | 0 |
| ARNTL(+) | 0 | 0 | 0 | 0 |
regulons <- regulonsToGeneLists(regulons_incidMat)
regulonAUC <- get_regulons_AUC(loom,column.attr.name='RegulonsAUC')
regulonAucThresholds <- get_regulon_thresholds(loom)
tail(regulonAucThresholds[order(as.numeric(names(regulonAucThresholds)))])
<dl class=dl-inline><dt>0.287117464540207</dt><dd>‘JUN(+)’</dd><dt>0.288565330122853</dt><dd>‘FOXP1(+)’</dd><dt>0.293575067056502</dt><dd>‘TAF7(+)’</dd><dt>0.312024320517666</dt><dd>‘KLF2(+)’</dd><dt>0.37288804895773</dt><dd>‘XBP1(+)’</dd><dt>0.440506596181331</dt><dd>‘HMGB1(+)’</dd></dl>
embeddings <- get_embeddings(loom)
close_loom(loom)
rownames(regulonAUC)
# names(regulons)
#### 2.导入seurat对象和加载的regulon信息进行匹配应对
sce <- readRDS(file = "../降维注释/data/pbmc注释后.rds")
# 取交集
sub_regulonAUC <- regulonAUC[,match(colnames(sce),colnames(regulonAUC))]
dim(sub_regulonAUC)
<ol class=list-inline><li>272</li><li>2638</li></ol>
sce
An object of class Seurat
13714 features across 2638 samples within 1 assay
Active assay: RNA (13714 features, 2000 variable features)
3 layers present: counts, data, scale.data
2 dimensional reductions calculated: pca, umap
#确认是否一致
identical(colnames(sub_regulonAUC), colnames(sce))
TRUE
# 构建细胞类型注释信息
cellTypes <- data.frame(row.names = colnames(sce),
celltype = sce$celltype)
head(cellTypes)
sub_regulonAUC[1:4,1:4]
| celltype | |
|---|---|
| <chr> | |
| AAACATACAACCAC-1 | Memory CD4 T |
| AAACATTGAGCTAC-1 | B |
| AAACATTGATCAGC-1 | Memory CD4 T |
| AAACCGTGCTTCCG-1 | CD14+ Mono |
| AAACCGTGTATGCG-1 | NK |
| AAACGCACTGGTAC-1 | Memory CD4 T |
AUC for 4 regulons (rows) and 4 cells (columns).
Top-left corner of the AUC matrix:
cells
regulons AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1 AAACCGTGCTTCCG-1
AHR(+) 0.00000000 0.000000000 0.032069971 0.040233236
APEX1(+) 0.14577259 0.232264334 0.072886297 0.187074830
ARID3A(+) 0.01093294 0.000000000 0.002935698 0.096007451
ARNTL(+) 0.02461937 0.005250513 0.013578447 0.002996437
table(sce$celltype)
# 根据自己需要的信息进行划分
selectedResolution <- "celltype"
cellsPerGroup <- split(rownames(cellTypes),cellTypes[,selectedResolution])
# 保留唯一/非重复的 regulon
sub_regulonAUC <- sub_regulonAUC[onlyNonDuplicatedExtended(rownames(sub_regulonAUC)),]
dim(sub_regulonAUC)
B CD14+ Mono CD8 T DC FCGR3A+ Mono Memory CD4 T
344 483 281 30 161 459
Naive CD4 T NK Platelet
721 145 14
272 2638
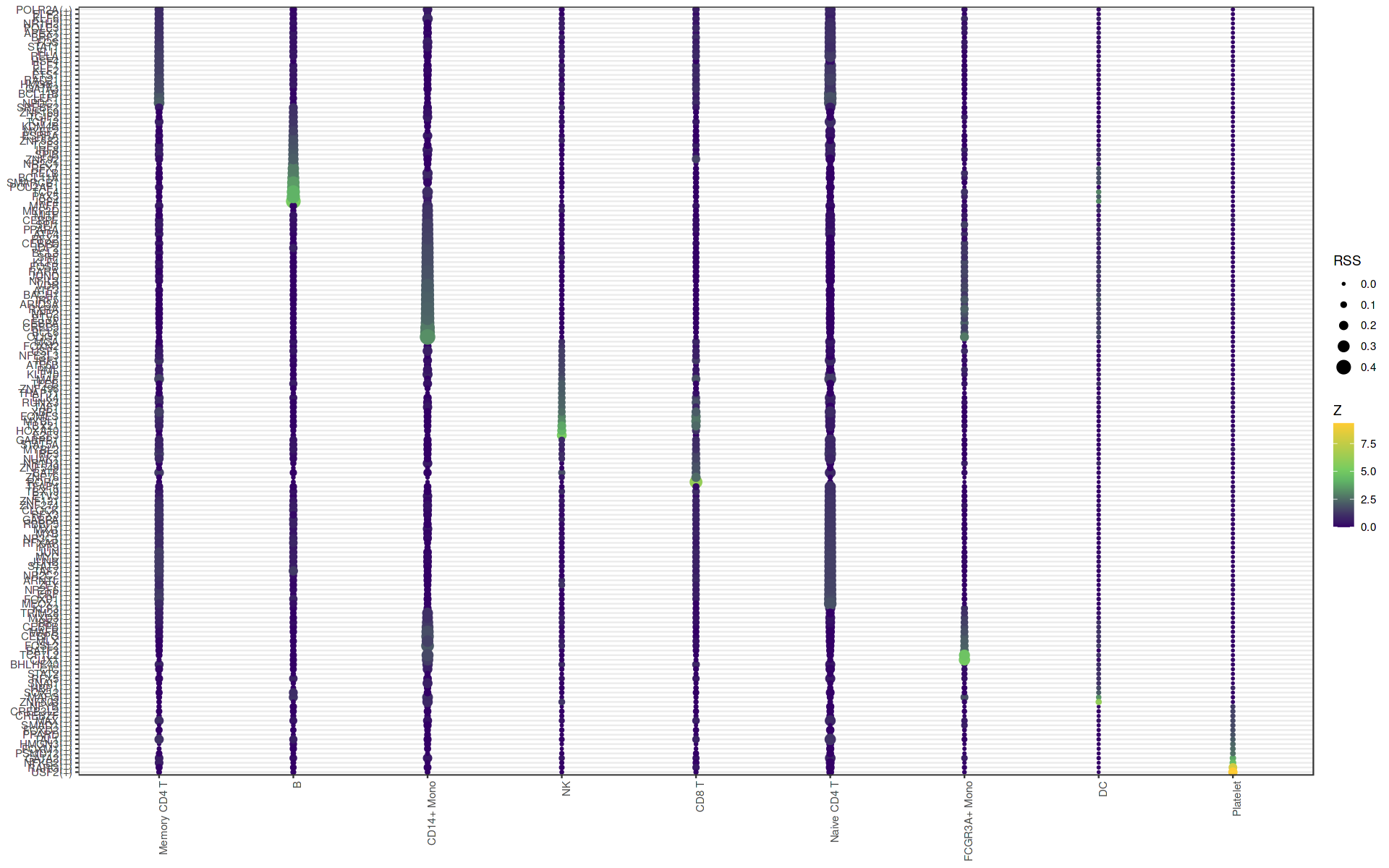
## 计算regulon特异性分数(RSS)
# regulon特异性分数(Regulon Specificity Score, RSS)
selectedResolution <- "celltype"
rss <- calcRSS(AUC=getAUC(sub_regulonAUC),
cellAnnotation=cellTypes[colnames(sub_regulonAUC),selectedResolution])
rss=na.omit(rss)
rssPlot <- plotRSS(rss,
labelsToDiscard = NULL, # 指定需要在热图中排除的行或列标签
zThreshold = 1, # 设定调控子的阈值,默认1
cluster_columns = FALSE, # 是否对列进行聚类
order_rows = T, # 是否对行进行排序
thr = 0.01, # 阈值参数,用于过滤 RSS 值。默认0.01
varName = "cellType",
col.low = '#330066',
col.mid = '#66CC66',
col.high= '#FFCC33',
revCol = F,
verbose = TRUE
)
rssPlot
options(bitmapType = "cairo")
p <- plotly::ggplotly(rssPlot$plot)
p <- p %>%
layout(
title = "RSS Plot",
xaxis = list(title = "Celltypes"),
yaxis = list(title = "Regulons")
)
p
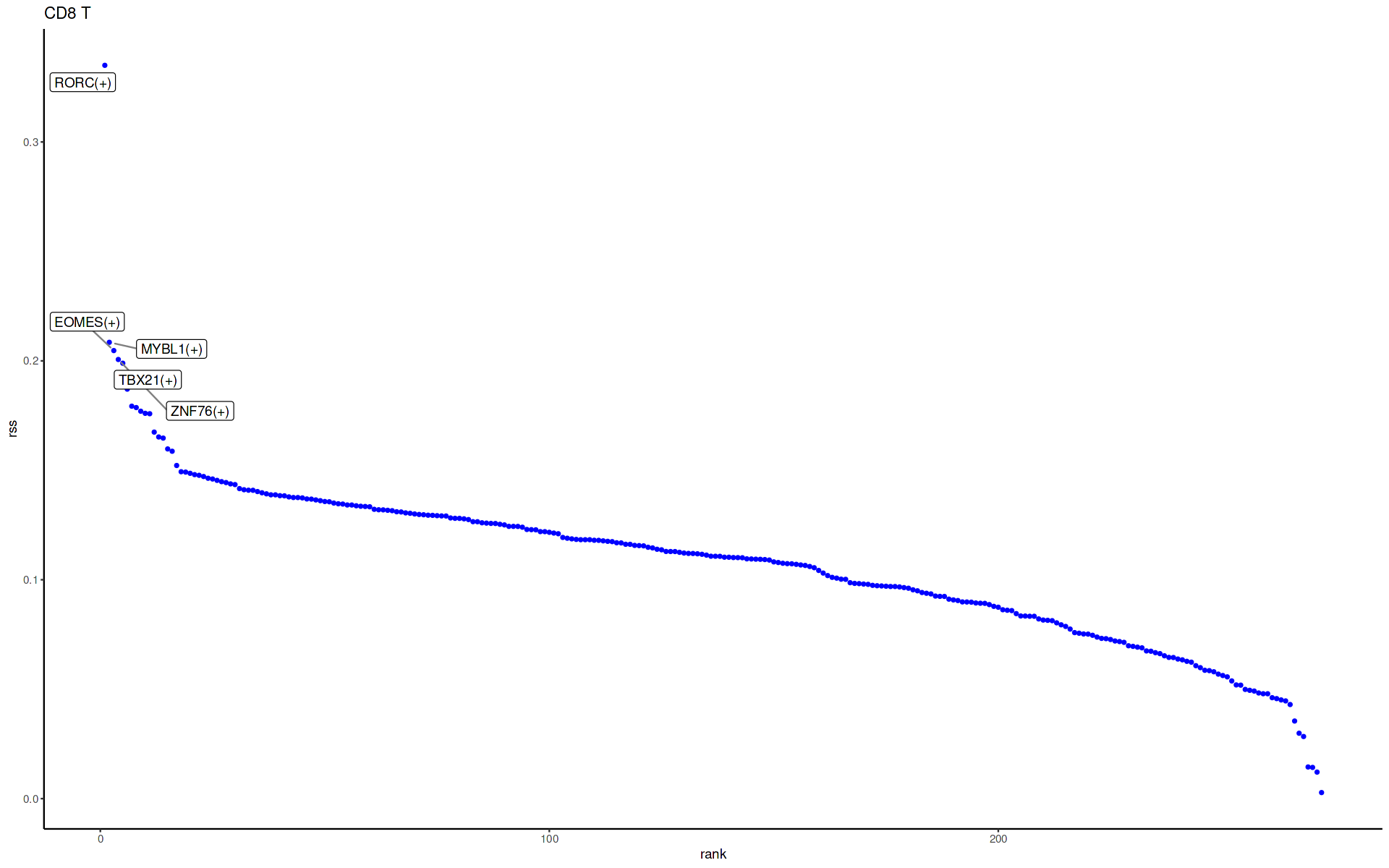
plotRSS_oneSet(rss, setName = "CD8 T") # cluster ID
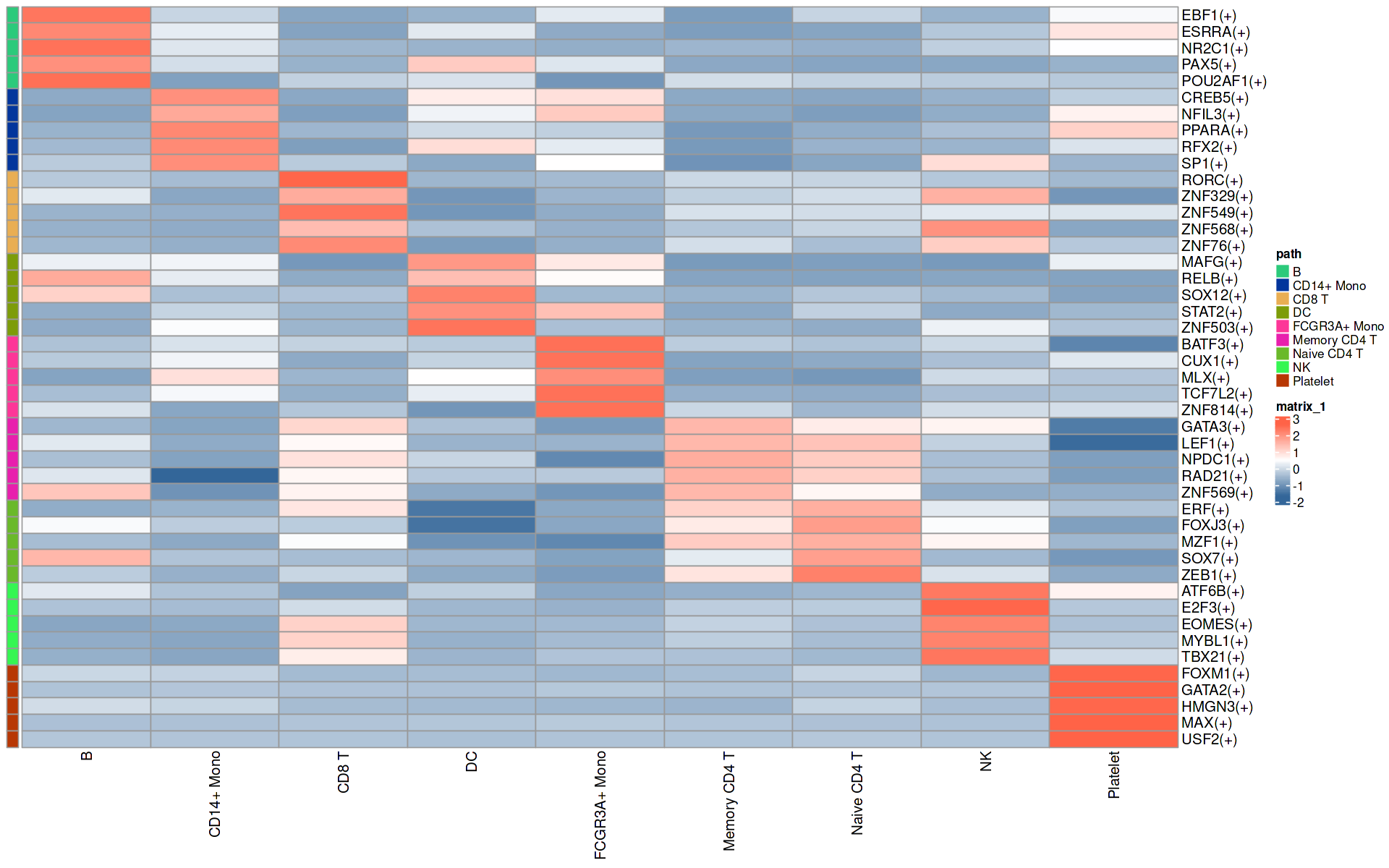
# 计算每个细胞组中各调控子(regulon)的平均活性,并将这些平均活性值存储在一个矩阵中
# cellsPerGroup这里得到是不同细胞群中的样本列表
# function(x)rowMeans(getAUC(sub_regulonAUC)[,x])可以计算每个细胞群的regulon平均AUC值
regulonActivity_byGroup <- sapply(cellsPerGroup,
function(x)
rowMeans(getAUC(sub_regulonAUC)[,x]))
range(regulonActivity_byGroup)
0.406250519055341
# 对结果进行归一化
regulonActivity_byGroup_Scaled <- t(scale(t(regulonActivity_byGroup),
center = T, scale=T))
# 同一个regulon在不同cluster的scale处理
dim(regulonActivity_byGroup_Scaled)
regulonActivity_byGroup_Scaled=regulonActivity_byGroup_Scaled[]
regulonActivity_byGroup_Scaled=na.omit(regulonActivity_byGroup_Scaled)
272
library(dplyr)
rss=regulonActivity_byGroup_Scaled
head(rss)
df = do.call(rbind,
lapply(1:ncol(rss), function(i){
dat= data.frame(
path = rownames(rss), # 当前regulon的名称
cluster = colnames(rss)[i], # 当前cluster的名称
sd.1 = rss[,i], # 当前cluster中每个调控因子的值
sd.2 = apply(rss[,-i], 1, median) #除了当前cluster之外的所有cluster 中该调控因子的中位值
)
}))
df$fc = df$sd.1 - df$sd.2
top5 <- df %>%
group_by(cluster) %>%
top_n(5, fc)
rowcn = data.frame(path = top5$cluster)
n = rss[top5$path,]
breaksList = seq(-1.5, 1.5, by = 0.1)
colors <- colorRampPalette(c("#336699", "white", "tomato"))(length(breaksList))
| B | CD14+ Mono | CD8 T | DC | FCGR3A+ Mono | Memory CD4 T | Naive CD4 T | NK | Platelet | |
|---|---|---|---|---|---|---|---|---|---|
| AHR(+) | -0.01079404 | 0.04430424 | -0.2551160 | -1.9998138 | -0.2291863 | 0.3974552 | -0.2097201 | 0.3835017 | 1.8793691 |
| APEX1(+) | 0.59661847 | -0.27602042 | -0.3590493 | 1.4097815 | 0.4591368 | 0.6754387 | 0.4772521 | -1.2602244 | -1.7229334 |
| ARID3A(+) | -0.66501429 | 1.40575469 | -0.8497800 | 0.7304750 | 0.7690242 | -0.7559845 | -0.8801283 | -0.9630327 | 1.2086858 |
| ARNTL(+) | -0.36067150 | -0.77655701 | 0.5700973 | -1.2267423 | -1.4662731 | 0.3939737 | 0.8414152 | 1.4448105 | 0.5799473 |
| ATF1(+) | 0.08143190 | -0.18468715 | 0.6530174 | -1.1488711 | 0.8302434 | 0.5776227 | 0.4197098 | 0.8450898 | -2.0735568 |
| ATF3(+) | -0.67963155 | 1.50856719 | -0.7699783 | 0.9926624 | 1.3901314 | -0.7276563 | -0.8960423 | -0.6147384 | -0.2033142 |
pheatmap(n,
annotation_row = rowcn,
color = colors,
cluster_rows = F,
cluster_cols = FALSE,
show_rownames = T,
#gaps_col = cumsum(table(annCol$Type)), # 使用排序后的列分割点
#gaps_row = cumsum(table(annRow$Methods)), # 行分割
fontsize_row = 12,
fontsize_col = 12,
annotation_names_row = FALSE)
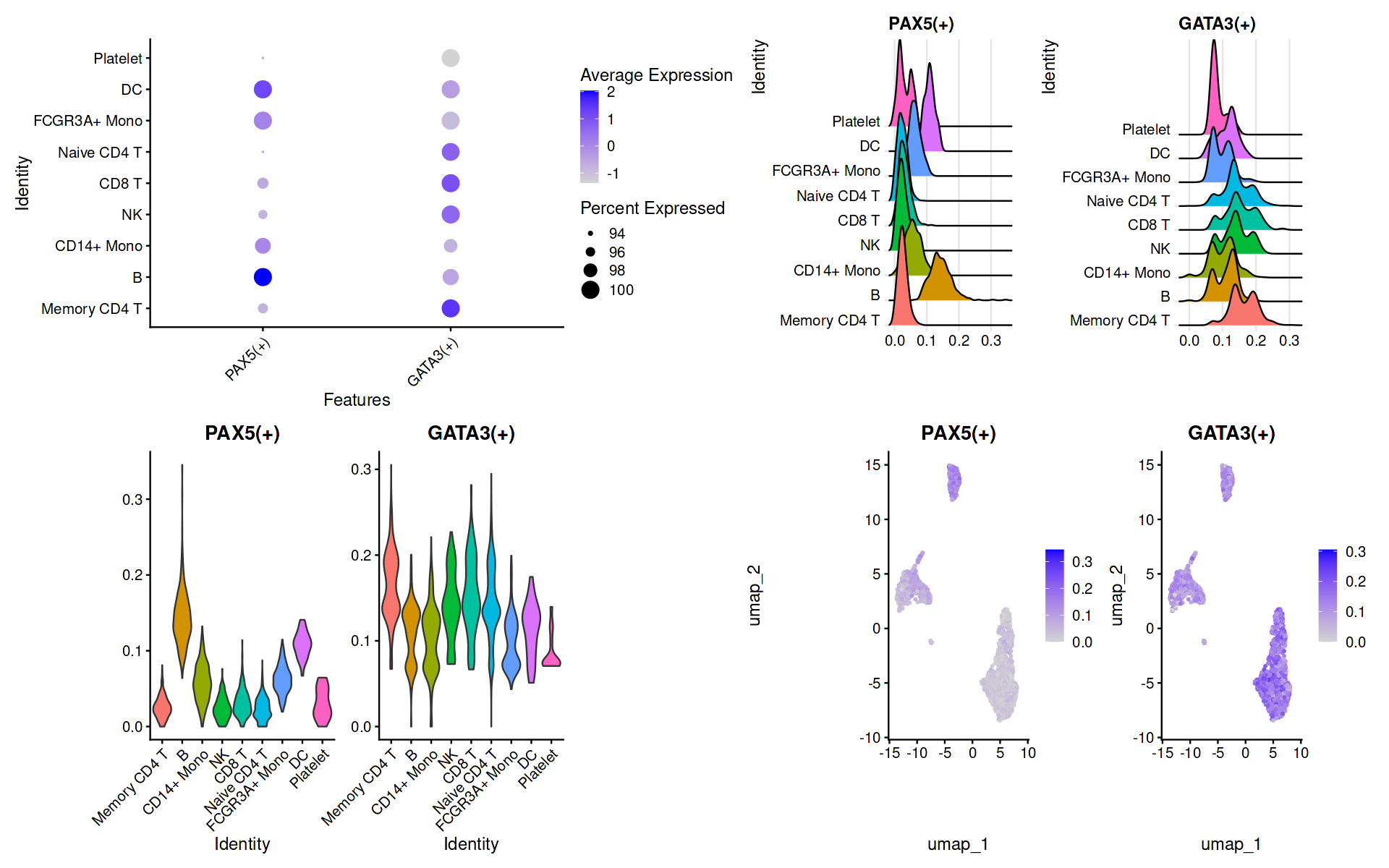
# 特定转录因子绘图
library(SummarizedExperiment)
seurat.data <- sce
Idents(seurat.data) <- "celltype"
regulonsToPlot = c("PAX5(+)","GATA3(+)")
regulonsToPlot %in% row.names(sub_regulonAUC)
seurat.data@meta.data = cbind(seurat.data@meta.data ,
t(assay(sub_regulonAUC[regulonsToPlot,])))
# Vis
p1 = DotPlot(seurat.data, features = unique(regulonsToPlot)) + RotatedAxis()
p2 = RidgePlot(seurat.data, features = regulonsToPlot , ncol = 2)
p3 = VlnPlot(seurat.data, features = regulonsToPlot,pt.size = 0)
p4 = FeaturePlot(seurat.data,features = regulonsToPlot)
wrap_plots(p1, p2, p3, p4, ncol = 2, widths = c(5, 5), heights = c(5, 5))
<ol class=list-inline><li>TRUE</li><li>TRUE</li></ol>
Picking joint bandwidth of 0.00626
Picking joint bandwidth of 0.0105